
Why Small Changes in Neural Transmission Affect Development
Easy Summary
Autism Spectrum Disorder (ASD) is a neurodevelopmental condition in which features appear in areas such as language, eye contact, and sensory processing due to differences in brain function. This article gently explains a brain substance called GABA (gamma-aminobutyric acid), which is thought to be related to the background of ASD.
GABA helps calm the brain’s neural excitability and allows information to flow smoothly. During a child’s brain development, GABA may temporarily act in the opposite way, supporting excitation. If this switch does not occur properly, traits associated with ASD are more likely to appear. Genetic factors (e.g., SLC12A5, MECP2) and the brain’s ion balance are also deeply involved.
In this article, we explain in simple terms the mechanisms related to ASD, common symptoms, methods of examination and diagnosis, treatment and support options, and future outlooks. We hope it will serve as a helpful guide for understanding and supporting children and their families.
Overview
GABA (gamma-aminobutyric acid) is widely known as the major inhibitory neurotransmitter in the mature mammalian brain. It calms neuronal activity and plays a critical role in ensuring that brain circuits form correctly and that information is transmitted properly.
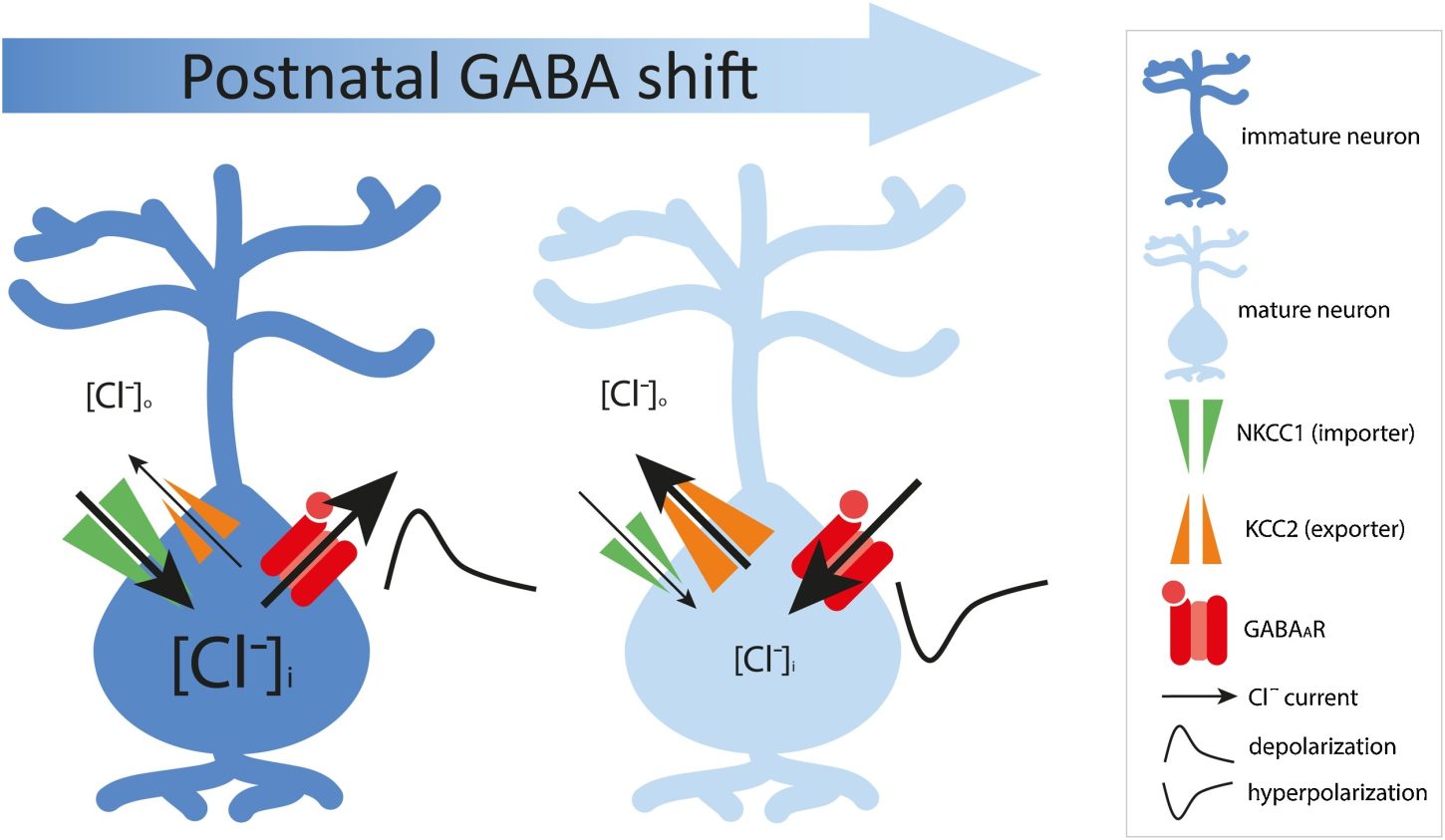
In the early stages of life, GABA signals are temporarily excitatory (depolarizing). This happens because the inside of neurons contains a high concentration of chloride ions (Cl⁻), maintained by the action of a chloride-importing transporter protein called NKCC1.
As the brain matures, expression of the chloride-exporting transporter KCC2 increases, reducing intracellular chloride levels. As a result, GABA signaling switches to inhibitory (hyperpolarizing). This switch, known as the postnatal GABA shift, is a crucial step in neural circuit development.
This polarity switch is not just a chemical change — it influences neuronal proliferation, differentiation, migration, synapse formation and pruning, network oscillations, and the balance of excitation and inhibition (the E/I balance).
When the timing, location, or degree of this GABA shift is disrupted, it has been linked to Autism Spectrum Disorder (ASD) and other neurodevelopmental disorders (NDDs).
Research in both ASD model animals and humans has shown that abnormalities in GABA function can manifest as either insufficient or excessive GABA activity. These abnormalities are often associated with genetic variations affecting GABA receptors, chloride transporters, and regulatory signaling factors.
Additionally, these GABA-related abnormalities intersect with the PI3K–AKT–mTOR pathway, a signaling pathway that regulates neuronal growth, metabolism, and plasticity. Dysregulation of this pathway is also implicated in the mechanisms underlying ASD.
Implicated Genomic Regions
Abnormalities in GABA signaling can arise from genetic variations in multiple loci. These genes encode subunits of GABA receptors, chloride co-transporters, synaptic proteins, and transcription factors.
For example, the 15q11–q13 region is frequently duplicated in ASD. This region contains genes such as GABRB3, GABRA5, and GABRG3, which encode subunits of the GABA<sub>A</sub> receptor, as well as UBE3A, which regulates synapse formation. Duplication of this region is associated with a subtype of ASD known as Dup15q syndrome.
Key chloride co-transporters KCC2 (SLC12A5) and NKCC1 (SLC12A2) also determine GABA polarity. Mutations or abnormal expression of these genes can prevent the developmental switch to inhibitory GABA, causing excessive neural excitation and aberrant circuit formation.
The PI3K–AKT–mTOR pathway also interacts with the GABA system. Genes such as PTEN, TSC1, TSC2, and NF1 regulate neuronal excitability, protein synthesis, and expression of chloride transporters.
Other ASD-associated genes such as MECP2, FMR1, SHANK1/2/3, CNTNAP2/3/4, and CDKL5 influence GABAergic synapse formation, interneuron function, and the E/I balance.
Additional relevant genes include GAD1/2 (for GABA synthesis), GPHN (for stabilizing synaptic structure), and NRXN1–3 (neurexins) and neuroligins for receptor localization and stability.
These genetic factors rarely act alone; they interact with epigenetic mechanisms, environmental influences, and sex-specific factors. For example, BDNF (brain-derived neurotrophic factor) and its receptor TrkB regulate KCC2 expression, a mechanism highlighted in both experimental and clinical studies.
Disorder
The primary condition associated with GABA dysfunction is Autism Spectrum Disorder (ASD), a highly heterogeneous neurodevelopmental disorder. Characteristics include difficulties with social communication and interaction, strong, focused interests, and repetitive behaviors.
Many individuals with ASD also experience comorbidities such as epilepsy, intellectual disability, ADHD, sleep disorders, or sensory sensitivities — all linked to imbalances in excitation and inhibition.
GABA dysfunction can be a cause or a consequence of these imbalances.
Syndromic forms of ASD with shared GABA pathway abnormalities include:
- Rett syndrome – caused by MECP2 mutations, often in females
- Fragile X syndrome – due to FMR1 dysfunction, with intellectual disability
- Angelman syndrome – caused by UBE3A abnormalities, leading to severe developmental delays
- CDKL5 deficiency disorder – associated with epilepsy and profound neurodevelopmental delay
- Dup15q syndrome – duplication of GABA receptor-related genes
- Down syndrome – trisomy 21, with reported NKCC1 expression abnormalities
- Dravet syndrome – SCN1A mutations impairing inhibitory interneurons, leading to epilepsy
These syndromes share disrupted GABAergic circuits, altered chloride homeostasis, and excessive network excitability, often intertwined with dysregulated PI3K–AKT–mTOR signaling.
Epidemiology
ASD is one of the most common neurodevelopmental disorders, affecting approximately 1 in 36 children worldwide.
A pronounced sex difference exists: males are diagnosed 3–4 times more frequently than females. Differences in the timing of GABA polarity shifts and transporter expression between sexes are thought to play a role. For instance, some studies suggest that females undergo the inhibitory GABA switch earlier than males.
Approximately 20–30% of individuals with ASD also have epilepsy, reflecting E/I imbalance.
Other frequent comorbidities include intellectual disability, ADHD, sleep disturbances, and sensory hypersensitivities.
Syndromic ASD, though rare, provides crucial insights into the shared molecular mechanisms — such as GABA pathway dysfunction and chloride imbalance — underlying ASD.
Etiology
The causes of Autism Spectrum Disorder (ASD) and related neurodevelopmental disorders (NDDs) are highly diverse and cannot be attributed to a single factor. Genetic mutations, environmental influences, and functional changes in neural circuits all interact at multiple levels.
Among these, two mechanisms have drawn particular attention: abnormalities in GABA function and disruptions of chloride homeostasis. These mechanisms, commonly observed across many cases, can be explained through two main axes:
1. Imbalance of Chloride Transporters and Failure of the GABA Polarity Shift
During fetal and early postnatal brain development, GABA normally acts as an excitatory (depolarizing) neurotransmitter, promoting neuronal growth and migration. This excitatory action occurs because of the high intracellular concentration of Cl⁻ maintained by the chloride importer NKCC1 (SLC12A2).
As development progresses, the chloride exporter KCC2 (SLC12A5) becomes more active, reducing intracellular Cl⁻ concentration. This allows GABA to switch to an inhibitory (hyperpolarizing) function — a critical turning point for the maturation of neural circuits, sensory processing, and behavioral regulation.
When this GABA polarity shift is delayed or fails to occur, GABA continues to excite neurons rather than inhibit them. This results in overexcitation and faulty neural connections, which can manifest as behavioral traits and sensory hypersensitivity commonly seen in ASD.
Conversely, premature inhibitory switching (such as through overexpression of KCC2 or early loss of NKCC1) can impair neuronal migration and network plasticity. Such abnormalities have been consistently observed in conditions including Rett syndrome, Fragile X syndrome, CDKL5 deficiency disorder, and in ASD models exposed to maternal inflammation or valproic acid during pregnancy.
2. Genetic and Molecular Abnormalities in GABA and mTOR Pathways
Many ASD risk genes are directly involved in the structure and regulation of the GABAergic system, including:
- GABA receptor subunits: GABRB3, GABRA5, GABRG3
- GABA synthesis and transport components: GAD1/2 (glutamic acid decarboxylase), VGAT (vesicular GABA transporter), gephyrin (receptor stabilizer)
- Chloride transporters: KCC2 (SLC12A5), NKCC1 (SLC12A2)
- Synaptic adhesion molecules: NRXN1–3 (neurexins), neuroligins (e.g., NLGN2)
- Transcriptional regulators: MECP2, TCF4, CHD8
A key point is that many of these genes are functionally linked to the PI3K–AKT–mTOR pathway.
- Genes like PTEN, TSC1/2, and NF1 act as negative regulators of the mTOR pathway. Mutations in these genes can lead to excessive protein synthesis and disrupted synaptic regulation.
- Genes such as SHANK3, UBE3A, and CHD8 regulate the structure and number of GABAergic synapses and influence mTOR signaling.
- MECP2, the gene implicated in Rett syndrome, modulates KCC2 expression via the BDNF–TrkB pathway, directly affecting the polarity shift of GABA.
Environmental factors such as valproic acid exposure during pregnancy, maternal immune activation, and oxidative stress have also been shown to disrupt GABA development, often through effects on chloride transporters and microglia.
Thus, GABA dysfunction may arise primarily from genetic causes or secondarily due to abnormalities in other neural networks. Overactivation of the PI3K–AKT–mTOR pathway has been reported to suppress GABA function and impair neural plasticity.
Symptoms
When GABA function is disrupted during critical periods of brain development, a range of symptoms common to ASD and other NDDs can emerge. These symptoms reflect not only local neuronal dysfunction but also widespread abnormalities in brain networks, oscillatory rhythms, and plasticity.
Core Behavioral Features
- Social communication difficulties
Disruption of E/I balance in the prefrontal and temporal cortices is implicated. Reduced activity of PV+ interneurons may impair neural synchrony needed for social information processing. - Restricted interests and repetitive behaviors
Abnormalities in GABA-A receptors or their transporters have been linked to similar behaviors in animal models. - Sensory hypersensitivity (especially tactile and auditory)
Altered GABA regulation of thalamocortical input can reduce the ability to filter sensory stimuli.
Neurological Comorbidities
- Epilepsy and seizures
Persistent excitatory GABA (due to inadequate chloride extrusion) can cause hyperexcitable neural networks, as seen in disorders such as Dravet syndrome and Rett syndrome. - Intellectual disability
Impaired GABA function disrupts synaptic pruning and network refinement, hindering cognitive development. - Sleep disturbances
Dysregulated GABA signaling in the hypothalamus and brainstem can alter circadian rhythms and sleep patterns.
Neurophysiological Characteristics
- Reduced gamma oscillations (30–80 Hz)
Reduced activity of PV+ interneurons leads to decreased gamma-band activity, associated with less efficient information processing. - Delayed or abnormal GABA polarity shift
This can lead to persistent giant depolarizing potentials (GDPs) and loss of network synchrony. - EEG abnormalities
Individuals with ASD often show irregular spectral power, especially reduced coherence in the gamma frequency range.
Sex- and Region-Specific Patterns
- Sex differences
Females tend to undergo the inhibitory switch earlier, possibly contributing to lower ASD prevalence in females. - Regional differences
Alterations in GABA markers and chloride transporter expression vary across brain regions, including the anterior cingulate cortex, hippocampus, and visual cortex.
In summary, these symptoms arise from a combination of timing mismatches in development and region-specific vulnerabilities, with circuit-level disinhibition as a central pathological feature.
Testing & Diagnosis
The diagnosis of Autism Spectrum Disorder (ASD) is still primarily based on clinical observation and behavioral assessment. Internationally, the DSM-5 (Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition) criteria are used, focusing on persistent difficulties in social communication and interaction, along with restricted and repetitive patterns of behavior, interests, or activities.
Recent research, however, is focusing on a deeper biological understanding of ASD to move toward precision medicine, exploring biomarkers for patient stratification.
Neuroimaging and Neurophysiological Biomarkers
- Magnetic Resonance Spectroscopy (MRS)
This non-invasive imaging technique allows for the quantification of GABA concentrations in specific brain regions. In ASD, studies have shown decreased GABA/glutamate ratios in areas such as the anterior cingulate cortex, prefrontal cortex, and somatosensory regions. However, results can vary depending on age and individual differences. - Electroencephalography (EEG)
EEG studies in ASD frequently reveal reduced gamma-band amplitude (30–80 Hz), altered spectral slopes, and increased variability in E/I balance. These patterns are being explored as potential biomarkers for stratifying ASD subtypes and predicting treatment responses. - Binocular Rivalry Paradigm
In visual perception tasks, correlations between GABA levels and the rate of perceptual switching are often disrupted, suggesting impaired inhibitory control in the visual cortex.
Anatomical and Genetic Findings
Postmortem studies provide critical insights into neural abnormalities in ASD. Findings include:
- Reduced parvalbumin-positive (PV+) interneurons
- Decreased expression of GABA-synthesizing enzymes GAD65 and GAD67
- Epigenetic suppression of KCC2 and certain GABA receptor genes
Although genetic testing is not yet routine for all ASD diagnoses, it can reveal causative factors in specific cases, such as:
- Single-gene mutations (e.g., MECP2, FMR1, SHANK3)
- Single nucleotide polymorphisms (SNPs) in GABA-related genes (e.g., GABRB3, GABRA5, SLC12A5)
- Structural chromosomal changes (copy number variants, CNVs) such as Dup15q syndrome
Such findings are increasingly valuable in identifying syndromic ASD and informing personalized treatment strategies.
Treatment & Management
At present, there are no medications that specifically target the core symptoms of ASD. Treatment is often focused on managing comorbid conditions like irritability, epilepsy, or sleep disturbances.
However, recent advances in understanding GABA dysfunction and chloride imbalance have driven the development of novel therapeutic approaches, ranging from experimental models to clinical trials.
1. Targeting Chloride Transporters
- Bumetanide
Bumetanide is a diuretic that selectively inhibits NKCC1, reducing intracellular Cl⁻ levels and promoting the developmental switch of GABA from excitatory to inhibitory.
- In animal models, bumetanide has improved both behavioral symptoms and neural network function.
- Small clinical trials in children have reported symptomatic improvements, particularly in those with abnormal EEG patterns or elevated inflammatory markers.
- Challenges include limited penetration across the blood-brain barrier and side effects like hypokalemia and dehydration, requiring careful monitoring.
- In animal models, bumetanide has improved both behavioral symptoms and neural network function.
- KCC2 Activators
Still in preclinical stages, drugs like CLP257 aim to enhance KCC2 activity, facilitating chloride extrusion and restoring inhibitory GABA signaling, with promising effects on synaptic plasticity and dendritic spine structure in models.
2. Modulating GABA Receptors
- GABA-A Receptor Agonists and Modulators
Compounds such as gaboxadol and benzodiazepine derivatives can enhance tonic inhibition, but responses are variable. Some individuals experience sedation, while others may see paradoxical increases in anxiety or hyperactivity. - GABA-B Receptor Agonists (e.g., Baclofen)
In animal models, baclofen has reduced repetitive behaviors and normalized neural oscillations, suggesting therapeutic potential.
3. Targeting Molecular Pathways
- PI3K–AKT–mTOR Pathway Modulation
In conditions like tuberous sclerosis complex (TSC) or PTEN-related ASD, mTOR inhibitors such as everolimus have shown benefits in restoring E/I balance and network synchronization. - BDNF–TrkB Pathway Regulation
Strategies to stimulate this pathway can enhance KCC2 expression, facilitating the GABA polarity shift.
4. Gene and Cell Therapies
- Gene Therapy
In Rett syndrome models, reactivation of the MECP2 gene has partially restored GABAergic transmission and improved behavioral outcomes. - Cell Therapy
Transplantation of interneurons derived from the medial ganglionic eminence (MGE) into ASD model mice has restored inhibitory function and improved social behaviors.
5. Non-Pharmacological Interventions
- Sensory Enrichment
In rodent models, enriched environments enhance KCC2 expression and improve inhibitory function. - Lifestyle and Neuromodulation
Dietary interventions, physical activity, and non-invasive brain stimulation techniques like transcranial direct current stimulation (tDCS) or transcranial magnetic stimulation (TMS) are being investigated for their ability to modulate GABA tone.
Future Directions
Research is focusing on:
- Using EEG gamma oscillations and cytokine profiles to stratify patients and predict treatment response
- Combining drug and non-drug interventions for synergistic effects
- Leveraging iPSC-derived neuronal models for personalized drug screening and precision therapy development.
Prognosis
The prognosis for individuals with ASD varies widely and depends on when and how GABAergic and broader neural circuit dysfunctions occur.
Factors Linked to Better Prognosis
- Early, appropriate interventions during critical developmental windows can promote circuit maturation and improve long-term outcomes.
- Absence of significant intellectual or language delays is associated with better social and adaptive outcomes.
- Identification of specific genetic alterations (e.g., imbalances in KCC2/NKCC1) can inform targeted treatments like bumetanide.
Factors Associated with Greater Challenges
- Significant delays or failures in the GABA polarity shift can lead to persistent circuit miswiring and reduced cortical plasticity.
- Persistent excitatory GABA, as seen in Rett syndrome or Dravet syndrome, often correlates with more severe symptoms.
- Adolescence and adulthood may bring additional changes due to hormonal shifts, environmental stress, or chronic inflammation, necessitating ongoing monitoring.
Neuroplasticity and Compensation
Despite early GABA dysfunction, the brain retains remarkable plasticity and adaptability. With experience and support, alternative neural pathways can form, enabling learning, social engagement, and functional adaptation.
Many individuals adapt over time, developing new strategies for communication and interaction. Supportive environments and timely interventions significantly enhance this adaptive capacity.
Conclusion
Understanding the mechanisms of a single neurotransmitter like GABA provides profound insight into the complexity of ASD and other neurodevelopmental disorders.
GABA receptors, chloride transporters, and signaling pathways such as PI3K–AKT–mTOR work together to maintain the delicate balance of excitation and inhibition in the brain. This balance can be disrupted by genetic mutations, slight developmental timing errors, or transient inflammatory responses.
Research shows that these imbalances do not represent a fixed fate but rather a dynamic interplay of vulnerability and resilience. Even when GABA remains excitatory or interneuron migration falters, the brain seeks alternative pathways and attempts to adapt.
This reality carries not only scientific significance but also a deeply human reminder: visible function does not always mean the absence of struggle, and the need for support cannot be judged solely by outward signs.
The nervous system, intricate and fragile yet resilient, orchestrates every moment of perception, learning, love, and meaning-making. Even when its balance wavers, possibility remains. Human development, in all its vulnerability and richness, deserves both understanding and support at every stage.
References
- Topchiy, Irina, et al. “GABA System as the Cause and Effect in Early Development.” Neuroscience & Biobehavioral Reviews, vol. 161, Jun. 2024, p. 105651. https://doi.org/10.1016/j.neubiorev.2024.105651.
- Bruni, Oliviero, et al. “Herbal Remedies and Their Possible Effect on the GABAergic System and Sleep.” Nutrients, vol. 13, no. 2, Feb. 2021, p. 530. https://doi.org/10.3390/nu13020530.
- Tang, X., Jaenisch, R. & Sur, M. “The role of GABAergic signalling in neurodevelopmental disorders.” Nature Reviews Neuroscience 22, 290–307 (2021). https://doi.org/10.1038/s41583-021-00443-x.
- Zhao, Haisheng, et al. “GABAergic System Dysfunction in Autism Spectrum Disorders.” Frontiers in Cell and Developmental Biology, vol. 9, Feb. 2022, p. 781327. https://doi.org/10.3389/fcell.2021.781327.
- Cherubini, Enrico, et al. “Dysregulation of GABAergic Signaling in Neurodevelopmental Disorders: Targeting Cation-Chloride Co-Transporters to Re-Establish a Proper E/I Balance.” Frontiers in Cellular Neuroscience, vol. 15, Jan. 2022, p. 813441. https://doi.org/10.3389/fncel.2021.813441.
- Ferranti, Anthony S., et al. “Novel Pharmacological Targets for GABAergic Dysfunction in ADHD.” Neuropharmacology, vol. 249, May 2024, p. 109897. https://doi.org/10.1016/j.neuropharm.2024.109897.
- Giniatullin, Rashid, and Enrico Cherubini. “GABAergic Signalling in Autism Spectrum Disorders (ASD): Role of Glial Cells and Therapeutic Perspectives.” Brain, Behavior, and Immunity, vol. 129, Oct. 2025, pp. 681–689. https://doi.org/10.1016/j.bbi.2025.07.003.
- Peerboom, Carlijn, and Corette J. Wierenga. “The Postnatal GABA Shift: A Developmental Perspective.” Neuroscience & Biobehavioral Reviews, vol. 124, May 2021, pp. 179–192. https://doi.org/10.1016/j.neubiorev.2021.01.024.
Helpful Terms
NKCC1 (SLC12A2)
A chloride importer abundant during fetal and early postnatal stages, maintaining excitatory GABA signaling in early development.
Autism Spectrum Disorder (ASD)
A condition where neural development follows unique patterns, leading to differences in social communication, sensory processing, and patterns of interests and behaviors. Symptoms and their intensity vary widely, as do intellectual and language abilities.
Neurodevelopmental Disorder (NDD)
A broad term for conditions related to brain development, including ASD, ADHD, and intellectual developmental disorders. They typically result from complex interactions between genetic and environmental factors.
GABA (Gamma-Amino Butyric Acid)
A neurotransmitter that calms neural activity. During early development, it can temporarily excite neurons but switches to an inhibitory role as the brain matures.
Inhibitory Neurotransmitter
A chemical that dampens neuronal activity, helping prevent overexcitation and maintain balance in the brain. GABA is the primary example.
Excitation/Inhibition (E/I) Balance
The balance between excitatory and inhibitory neural signals. Disruptions in this balance can lead to sensory hypersensitivity, sleep disturbances, and seizures.
GABA Polarity Shift
The developmental change where GABA switches from excitatory to inhibitory. Delays or disruptions in this shift can affect neural circuit formation and contribute to atypical development.
Chloride Transporter
Proteins that regulate the movement of chloride ions in and out of neurons. KCC2 expels chloride, while NKCC1 imports it, influencing GABA polarity.
KCC2 (SLC12A5)
A chloride exporter critical for the shift of GABA to an inhibitory role during brain maturation.
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。







