







お盆期間(8月13日〜15日)も通常どおり診療しております
- 知る権利の尊重
- 妊婦さんが安心して健やかな出産にのぞめるように、妊婦さんとご家族の”知る権利”を尊重します。
- 正しいNIPTの理解へ
- NIPT(新型出生前診断)は胎児の健康状態を知るだけでなく、母体の健康維持に繋がると広く理解されることを目指します。
- IT × 医療
- ITの活用により妊婦さんの利便性の向上、検査精度の向上、迅速な対応を行ないます。



ヒロクリニック利用者を対象に「2023年12月NIPT(新型出生前診断)に対する満足度」に関するアンケート調査を実施しました。
2023年12月 株式会社イージークラウド調べ


ヒロクリニックの関連会社である
「東京衛生検査所」は、もともと認証施設としてNIPTを提供してきました。
その経験を踏まえ、より多くの妊婦さんのニーズに応えられる独自の検査体制へ移行していますが、検査内容は同じです。
認証施設では学会基準に基づき限られた染色体異常を対象とした検査が行われますが、 ヒロクリニックではその知見を活かし、染色体の数の異常だけでなく、知的障害を伴う疾患の原因となることがある染色体の微細な欠失や重複など、より幅広い遺伝学的変化にも対応できる検査プランを提供しています。
また、受検条件の柔軟さや受診のしやすさにも配慮し、検査の選択肢を広げた医療サービスを目指しています。
認証施設を運営してきたヒログループ。
その経験と知見を受け継ぎ
さらに進化したNIPTへ。



NIPT(新型出生前診断)とは、お母さんの腕から採血するだけで、おなかの赤ちゃんにダウン症などの染色体の病気の可能性があるかを、高い精度で調べる出生前検査です。採血のみで赤ちゃんに負担がなく、流産のリスクがありません。結果は「可能性」を示すもので、診断の確定には羊水検査が必要です。
| 検査方法 | お母さんの採血のみ(おなかに針を刺さず、流産リスクなし) |
|---|---|
| 受けられる時期 | 一般に妊娠10週0日ごろ〜。ヒロクリニックは心拍確認後の早期から受検可能(年齢制限なし・紹介状不要) |
| 精度 | 感度99.9%以上/陰性的中率99.99%(対象疾患) |
| 結果までの日数 | 最短2〜5日(国内の自社ラボ「東京衛生検査所」で解析) |
| 主にわかること | 13・18・21トリソミー(ダウン症など)をはじめとする染色体疾患。プランにより全染色体や微小欠失なども対象 |
| 費用の目安 | 基本プランで66,000円(税込)〜。検査範囲・プランにより異なります |
| 検査の位置づけ | 非確定的検査(スクリーニング)。陽性時は遺伝カウンセリングのうえ羊水検査などの確定検査を検討 |
※料金・プランの詳細は料金プランページを、NIPTの詳しい解説はNIPT(新型出生前診断)とはをご確認ください。
陽性スコアは、カットオフ値※からどれだけ離れているかを示す数値で、数値が大きいほど陽性である可能性が高くなります。 羊水検査を行う前の判断材料の一つとして活用できます。
※カットオフ値:検査結果を陽性か陰性かに分ける基準となる数値
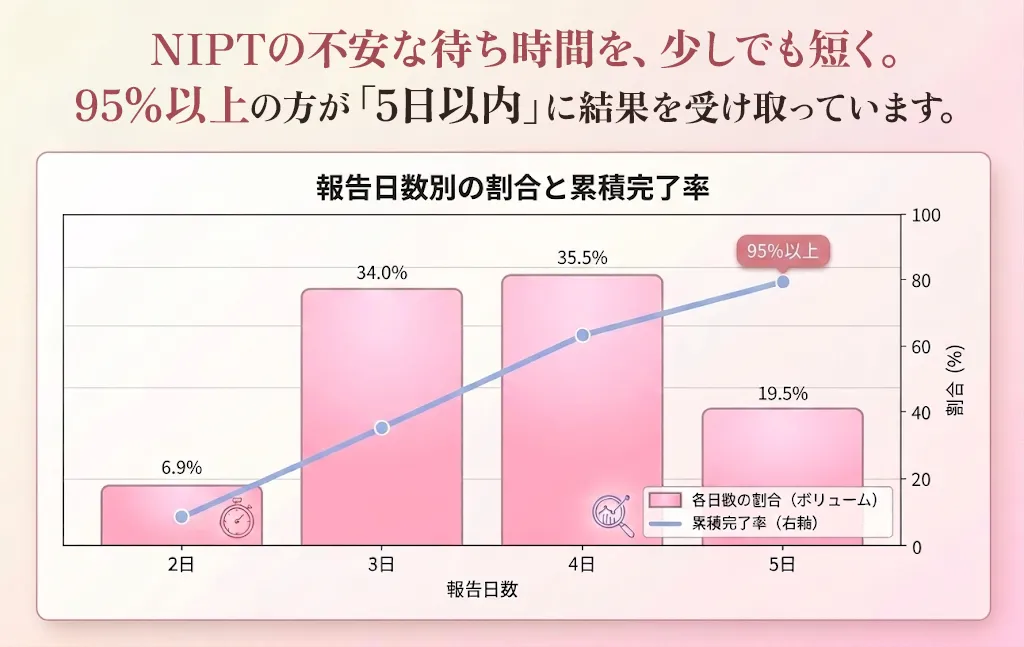
実績75,000件以上!
NIPTだから結果が早く、確実で安心。
特急便なら最短2日。95%が5日以内で届きます。
ご予算やニーズに合わせて必要な検査だけを選択可能。
母体・胎児へのリスクはゼロ。
週数や年齢による制限もありません。
土日祝対応・お一人での来院もOK!
お近くのクリニックで気軽に受診できます。
全染色体・微小欠失症・性別検査も可能。
※1 2026年6月時点の実績
※2 直営院10院+連携クリニック115院
2026年6月調べ




岡 博史先生
岡山県出身
医療法人社団福美会 ヒロクリニック統括院長/Labo Director
慶應大学医学部卒業後、日本とアメリカの医師国家試験を合格。臨床研修後に2年で医学博士を取得。
専門職大学の客員教員就任やNIPT専門クリニックの医学博士として活躍。日本で20人ほどしか存在しないラボディレクターの資格を有している。
日本で行われているNIPTはさまざまな制限を受けており、利用者が結果的に必要なものを受けれていない現状に関して、世界最高峰のNIPTを提供したいとの思いでやっています。
お盆期間(8月13日〜15日)も通常どおり診療しております
ARTICLES
知的障害のあるお子さまの将来に不安を感じるのは、親としてごく自然なことです。
「この子は大人になったら仕事ができるのか…」
「親がいなくなったあと、この子はどうやって生きていけるのか…」
そうした思いに、私たちも日々向き合っています。
こうした知的障害とはどのくらいあるのでしょうか?
知的障害の細かい情報はこちらのページに記載してありますが、結論からすると全ての知的障害をNIPTで検査できるわけではありません。
なぜなら知的障害は遺伝子と関係なく発症することのほうが多いからです。しかし、生まれる前にできる限りその可能性を除きたいと思うのは自然な考えだと思います。
一番多くの施設で行われている3種類のNIPT検査(13,18,21番染色体検査)では知的障害を検出することは可能でしょうか?
21番染色体の検査(ダウン症検査)のみが知的障害の検査の対象となります。まだ13番と18番トリソミーの検査に関しては身体的障害が重すぎて、知的障害の認知をすることが難しい状態です。つまり早番なくなってしまうために、知的障害であるかどうかもわからない位重症であると言うことです。
しかし、ご両親が1番心配するのは、身体的には成長するが、知的障害を残したたまま子供が成長することでは無いのでしょうか?
これが出生前にわかるとしたならば、できる限り、多くの疾患を検出したいと思うのが親心だと思います。では知的障害とはどのような場合に発症することがあるのかを考えてみたいと思います。
先ほど述べた13,18番トリソミーの場合、知的障害が発症しないのは、これらの染色体にあまりにも多くの重要な遺伝子が載っているために、知的障害のみといったようなケースとなる事は少ないのです。生命に関係するような遺伝子も多数載っているため、知的障害以前に生命を維持することが困難になってしまうのです。では一部の遺伝子の数または配列に異常があった場合、どうなるかと言うと生命維持はできるけれども、高度機能である脳の障害を伴い知的障害を起こすことが多いです。
NIPT検査は基本的に染色体の量を調べる検査です。染色体の配列異常を調べる事はできないため、NIPT検査できるのは、あくまでも染色体の部分的な量の変化です。遺伝子異常で最も多いのは配列の異常よりも量の異常です。そのためNIPT検査はここの量の異常を調べることができるためにとても有用です。
では、どのようにしたら、このような知的障害の検査ができるのでしょうか?
まずこのホームページを見られた方は、NIPT検査には複数の選択肢があると言うことに気づかれたと思います。産婦人科のドクターが勧める13, 18, 21番染色体以外の検査が存在すると言うことです。このそれ以外の検査と言うのは世界では広く行われてきております。特に有名なところでは性染色体の異常です。また一部の染色体かける微小欠失・重複疾患も有名な疾患です。なぜならこの中に含まれるディジョージ症候群は、知的障害の中では2番目に多い疾患と言われています。これはダウン症の次に多い知的障害です。その平均年齢は50歳と言われています。先ほどお話しした通り、一部の染色体が欠けた場合には生命維持することが可能なため、現在の医療では50代から60代といった寿命を持つことがあります。しかしながらその知能はIQ 50程度であり、1人で生活していくのには厳しいIQとなっております。また性染色体の異常はそのお子様の不妊、成長障害などを伴います。早期知っておくことによって成長障害の予防を行うことができ、また本人が自分が不妊症である確率が高いと言うことを知ることによって、TSTEなどの先進的な医療を受けることも可能となります。性染色体の異常に関しては、知的障害と呼ぶほどでは無いものも、学習障害は一般的に伴うことが多いです。これらの細かい情報に関しては、各ページに書かれてありますのでご覧ください。
この場で申し上げたい事は、NIPT検査は3種類だけではなく、現在では様々な検査ができるようになり、その多くは治療が完全に不可能な知的障害・発達障害を伴う疾患も早期に検出することが可能となっています。このような事実は、妊婦さんに刺されることなく、NIPT検査が徐々に広がってきていることに私は一定の不安を抱いております。
このホームページの中に詳しく書かれていますので、ご覧ください。文章だけではわかりにくい場合にはYouTubeの方を毎日提供しております。こちらも参考にしてください。
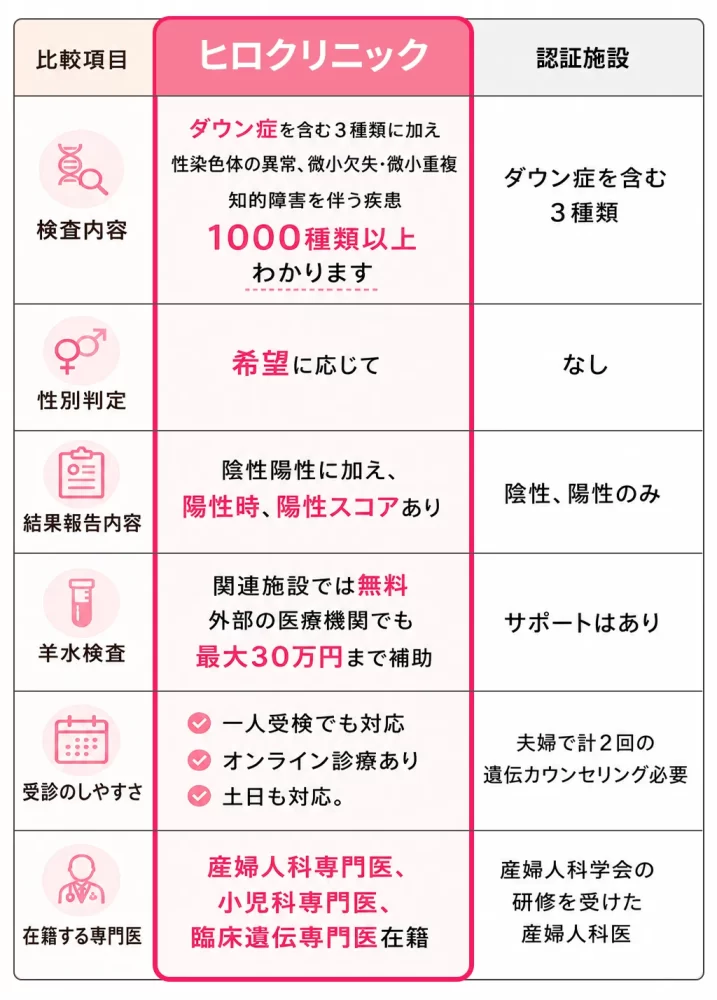
ヒロクリニックと他院(主に日本医学会が認定する認可施設)との主な違いは、「検査の自由度」と「受診のしやすさ」にあります。
まず検査項目ですが、認可施設では21・18・13トリソミーの3種類に限定されるのが一般的です。対してヒロクリニックは、全染色体や微小欠失、性別判定まで幅広く対応しています。
受診条件も異なります。認可施設は「35歳以上」などの年齢制限や紹介状を求める場合がありますが、ヒロクリニックは年齢制限がなく、心拍確認後という早期から受診可能です。さらに、国内検査所との連携により最短2〜5日で結果が分かるスピード感や、6万円台から選べる費用面も大きな利点です。
一方で、認可施設は対面での手厚い遺伝カウンセリングが必須であり、体制が厳格に管理されている安心感があります。詳細はヒロクリニック公式サイトの比較ページで確認し、自身の希望に合う方を選びましょう。
ヒロクリニック(HIRO CLINIC)は、主にNIPT(新型出生前診断)を専門に行う医療機関です。この検査では、お母さんの血液からお腹の赤ちゃんの染色体異常を高い精度で調べることができます。
具体的には、以下の内容を把握することが可能です。まず、21トリソミー(ダウン症候群)、18トリソミー、13トリソミーといった主要な染色体疾患の可能性が分かります。さらに、選択するプランによっては、1番から22番までのすべての常染色体や、染色体の一部が欠けている「微小欠失・重複疾患」の状態まで網羅的に検査できます。加えて、性染色体の異常や、赤ちゃんの性別についても判定が可能です。
国内の複数拠点で展開しており、年齢制限がなく、心拍確認後から受診できる点が大きな特徴です。詳細はヒロクリニック公式サイトで確認の上、検討してみてください。
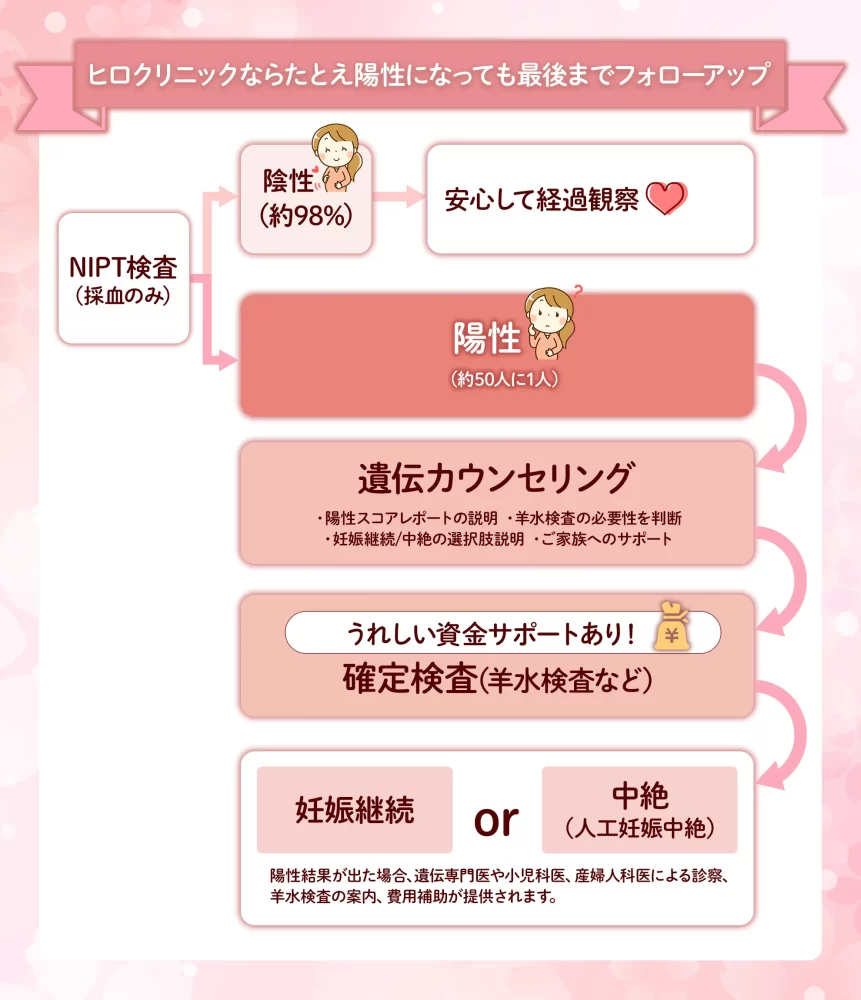
ヒロクリニックでNIPTの結果が陽性だった場合は、診断確定のために「羊水検査」を受けることが推奨されます。羊水検査には通常10万〜20万円ほどの費用がかかりますが、ヒロクリニックでは受検者の負担を軽減するために3つの「羊水検査サポート」を用意しています。
具体的には、3,300円(税込)のサポート料で最大10万円まで補助される「ライト」、5,500円(税込)で最大20万円までの「スタンダード」、そして11,000円(税込)でエコー代等の諸経費含め最大30万円まで補助される「ワイド」から選択可能です。お支払いは初回時の1回のみで、早期NIPT(Early NIPT)を受検された場合も追加費用なしで継続してサポートが適用されます。
本制度は他院での検査も対象となるほか、陽性判明後には的中率を数値化した「陽性スコア」レポートの提供や専門医への相談体制も整っており、落ち着いて今後の判断ができるよう手厚く支援しています。
詳細はヒロクリニックの陽性後のアフターケアをご確認ください。
ヒロクリニックの検査が早い主な理由は、国内の検査機関(東京衛生検査所)で検査しているからです。
多くの医療機関では、採集した検体を海外の検査機関へ輸送するため、結果が出るまでに1〜2週間ほどかかります。一方、ヒロクリニックは国内で検査を完結させ、さらに自動分注機や最新の次世代シーケンサーを導入して効率化を図ることで、最短2〜5日というスピード通知を実現しています。
精度については、他院や認可施設と比較して遜色ありません。NIPT自体の精度は非常に高く、陰性的中率は99.99%に達します。
ヒロクリニックにおいて、胎児のDNA不足などで結果が出ず「判定不能(判定保留)」となり、再検査が必要になる確率は非常に低く、約0.3〜0.4%程度とされています。
ヒロクリニックのNIPT検査は非常に高い「結果報告率」を誇り、実績データでは99.98%の人に何らかの結果が届けられています。判定不能になる主な原因は、お母さんの血液中に含まれる胎児由来のDNA量(胎児分画:FF)が基準を満たさないことです。特に妊娠週数が早い場合や、お母さんの体重、体質によってDNA濃度が薄くなることがありますが、その場合は1〜2週間ほど期間を空けて再採血することで、85%以上のケースで正しく判定が可能になります。
万が一、再検査でも判定ができなかった場合には、状況に応じて医師と今後の進め方(羊水検査への移行など)を相談する形となります。詳細はヒロクリニックの判定不能に関する解説ページで確認できます。
ヒロクリニックでは、妊娠週数が「早すぎる」場合でも「遅すぎる」場合でも、NIPT検査を受けることが可能です。
まず「早すぎる」ケースについてですが、一般的なクリニックでは妊娠10週からの受診が標準的であるのに対し、ヒロクリニックではエコーで胎児心拍が確認できれば受診できます。早期に受検することで、万が一陽性だった際にも、その後の精密検査や意思決定に十分な時間を確保できるのが大きな利点です。
次に「遅すぎる」ケースについても、受診の上限週数に制限はありません。妊娠15週以降や、18週を過ぎた「Late NIPT」にも柔軟に対応しており、妊娠後期に不安が生じた方でも検査を受けられます。
ただし、陽性時の確定診断となる羊水検査を受けられる時期には法的な制限や医学的な期限があるため、余裕を持って14週頃までに受診することが推奨されています。
ヒロクリニックでは、NIPTと胎児ドック(精密超音波検査)を検討される際、基本的には「まずNIPTを受けること」を推奨しています。
NIPTは採血のみで受検でき、染色体異常の可能性を極めて高い精度で判定可能です。まずNIPTで全体のリスクを確認し、陰性であれば安心してその後の妊婦健診へと戻ることができます。もし陽性や判定保留となった場合には、次のステップとして胎児ドックや羊水検査へ進むという流れが、医学的にも合理的です。
先に胎児ドックで異常を指摘されてからNIPTを受けるケースもありますが、NIPTの結果を基に専門的なエコーで詳細を確認する方が、診断の効率が良く、妊婦様の精神的負担も抑えられると考えられています。
2026年現在、当院ではNIPTと超音波検査を組み合わせたプランも提供しております。
最適な受検時期を医師へご相談ください。
当院の支払い方法と、当日の所要時間についてご案内いたします。
【お支払い方法について】
2026年現在、当院では患者様の利便性を考慮し、多彩な決済方法を導入しております。現金払いはもちろん、主要なクレジットカード(VISA、Mastercard、JCB、American Express、Diners等)がご利用いただけます。また、各種QRコード決済や電子マネーにも対応しておりますので、ご自身に合った方法をお選びください。
【当日の所要時間について】
ご来院からお帰りまでは、通常30分〜1時間程度と非常にスムーズです。事前予約制のため待ち時間が少なく、当日は問診票の記入、医師によるカウンセリング、採血という流れで進みます。お仕事や育児の合間にも無理なく受診いただける体制を整えております。
当院の支払い方法と、当日の所要時間についてご案内いたします。
【お支払い方法について】
2026年現在、当院では患者様の利便性を考慮し、多彩な決済方法を導入しております。現金払いはもちろん、主要なクレジットカード(VISA、Mastercard、JCB、American Express、Diners等)がご利用いただけます。また、各種QRコード決済や電子マネーにも対応しておりますので、ご自身に合った方法をお選びください。
【当日の所要時間について】
ご来院からお帰りまでは、通常30分〜1時間程度と非常にスムーズです。事前予約制のため待ち時間が少なく、当日は問診票の記入、医師によるカウンセリング、採血という流れで進みます。お仕事や育児の合間にも無理なく受診いただける体制を整えております。
当院では10週から14週を推奨しております
お受けいただけます。
何週目までに受けなければならないという制限はありません。ただ、羊水検査などを考慮しても、早めが望ましいです。
1回目の採血から2週間後かつ10週0日以降14週までの期間を推奨しております。
可能性はございます。
再採血を無料で行っております。


Copyright (c) NIPT Hiro Clinic All Rights Reserved.
 AIチャット
AIチャット
