
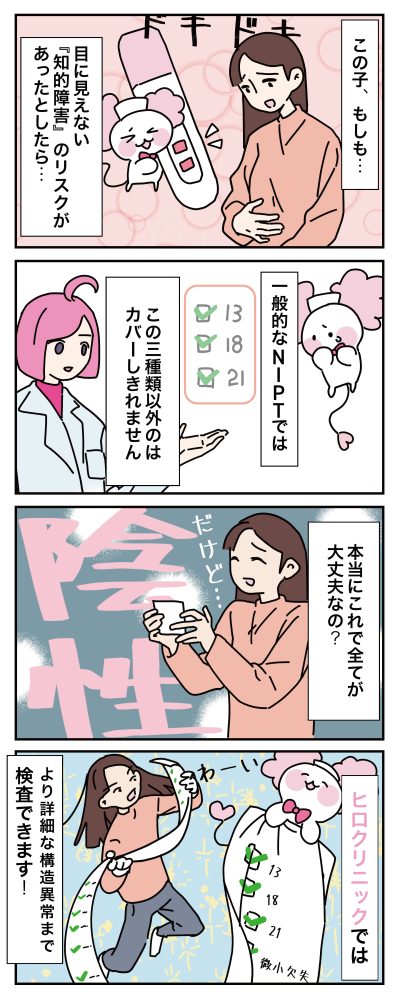
NIPTy Standardってなに?
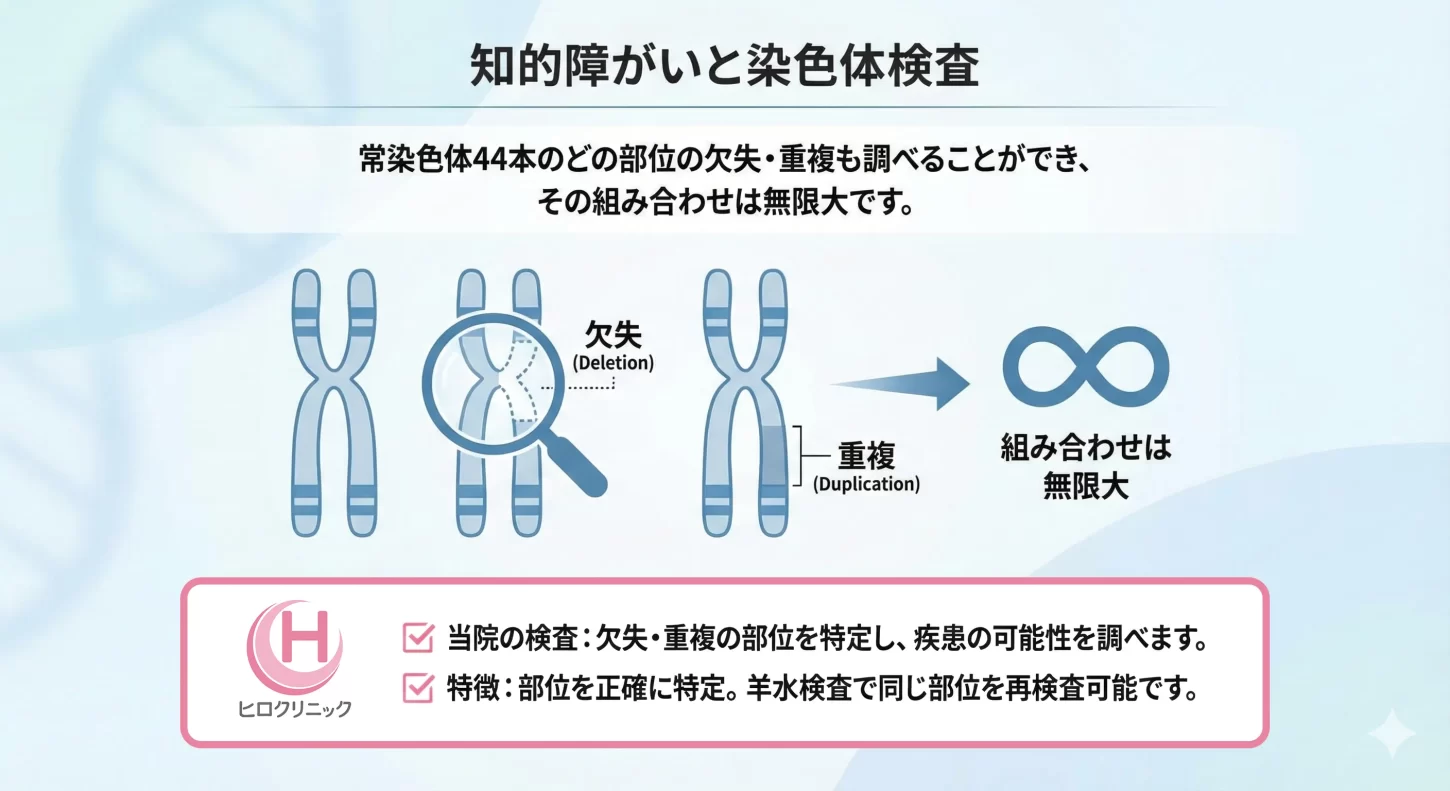
NIPTy Standardでは、常染色体44本のどの部位の欠失・重複も調べることができ、その組み合わせは無限大です。
この検査では、欠失・重複がどの部位にあるかを特定し、疾患の可能性について調べます。下部に記載されている代表的な組み合わせもありますが、実際の欠失・重複の組み合わせ数は無限大です。ヒロクリニックでは、欠失・重複の部位を正確に特定し、羊水検査で同じ部位を再検査することが可能です。

検査可能プラン
ヒロクリニックが検査を依頼している東京衛生検査所の次世代シーケンサーでは、500万塩基以上の欠失・重複が検出された場合に、検査結果として報告しています。
微小欠失疾患は、100万塩基から500万塩基程度の欠失でも症状を発症することがありますが、症例によっては500万塩基以上の欠損を伴うこともあります。疾患特有の遺伝子欠損を超える大きな欠失があった場合でも、その遺伝子が欠損しているため、同様の症状やより重度の疾患を引き起こす可能性があります。このため、欠損部位が疾患特有の部位に含まれている場合、欠損範囲が広くても、同じ診断名を付けることが妥当だと考えています。微小欠失と呼ぶと誤解を招く恐れがあるため、当サイトでは「部分欠失疾患」として記載しています。
欠失・重複は、その部位に存在する遺伝子がどのような働きを持っているかによって、さまざまな症状を引き起こします。例えば、ある酵素を作り出す遺伝子を含む染色体が欠損すると酵素欠損症が発生し、骨形成に関わる遺伝子を含む染色体が重複すると骨形成異常が見られます。
従来、染色体の欠失・重複は、染色体を染色し顕微鏡下で検出していましたが、この方法では1,000万塩基(10Mb)以上の欠失・重複しか検出できませんでした。近年、次世代シーケンサーやマイクロアレイ染色体検査を用いることで、より精度の高い検査が実施できるようになり、欠失・重複は特定の染色体に限定されず、全染色体にわたって発生することが明らかになりました。これにより、欠失・重複と、それに伴う臨床的所見との関連が報告され、異常の認められた遺伝情報がデータベース化されています。このデータベースには、共通の異常領域と特徴的な所見に基づく複数の報告例が含まれており、稀な例や新たに報告されたケースもあります。
したがって、独立した疾患として報告されていない欠失・重複がある場合、症例報告やゲノムデータベースを参照して症状などを調査する必要があります。東京衛生検査所では、染色体の部分的な欠失・重複が検出された際に、国内外の信頼性の高いデータベースを検索し、該当する疾患情報を担当医師に提供しています。
すべての重複領域における遺伝子と臨床所見との関連性は、まだ明確にはなっていません。また、欠失・重複が認められた場合でも、症状が現れないことがあります。これは、欠失・重複する部位に生命活動や身体の形成に関わる重要な遺伝子が存在しない場合、または遺伝子が存在していても病的所見を示さない場合があるためです。例えば、8番染色体の短腕(8p23.2)の重複は、250万塩基(2.5Mb)に及ぶものでありながら、がん抑制遺伝子のみが含まれているため、異常所見は認められず、正常変異として理解されることもあります。
欠失・重複が認められた場合、その病的な影響を鑑別する必要がありますが、これまでに報告されたことのないケースについては、その判断が難しいことや、欠失・重複との関連が不明な場合(variant of unknown significance: VUS = 意義不明変異)があることをご理解ください。
名もなき症例
全染色体部分欠失・重複疾患で近年になって報告された症例の論文の一例です。
| 染色体 | 染色体座位 | 論文 |
|---|---|---|
| 2番 | 2p16.3-p21 | 2番染色体(2p16.3‐p21)の隣接遺伝子欠失:リンチ症候群の原因として(翻訳) |
| 7番 | 7q21.3-q31.1 | 7番染色体(7q21.3-q 31.1)部分重複: まれな分節性ゲノム異数性: 症例報告と遠位および類似の分節が関与した症例のレビュー(翻訳) |
| 12番 | 12q24.31-q24.33 | 12番染色体(12q24.31-q24.33)部分欠失に伴う自閉症:非常に稀な疾患の追加報告(翻訳) |
| 14番 | 14q | 14番染色体の部分重複(翻訳) |
| 19番 | 19p13.13 | 19番染色体(19p13.13)の新規微小欠失/微小重複症候群(翻訳) |
全染色体部分欠失・重複疾患の報告例
| 染色体 | 欠失部 | 症候群 | 備考 |
|---|---|---|---|
| 1番 | 1p12 | アラジール症候群(Alagille症候群) | |
| 1q21.1 | 1q21.1微細欠失症候群 | ||
| 1p36 | 1p36欠失症候群 | 発生頻度(出生時)4,000〜10,000件中1件 成長障害、重度精神発達遅滞、難治性てんかんなどの症状 |
|
| 2番 | 2q13 | ネフロン癆(Nephronophthisis)1型 | |
| 2p21 | 全前脳胞症 | ||
| 2q37.3 | オルブライト(Albright)症候群様中手骨・中足骨短縮 | ||
| 3番 | 3q29 | 3q29微細欠失症候群 | |
| 4番 | 4p16.3 | ウォルフヒルシュホーン症候群(Wolf-Hirschhorn症候群) | 発生頻度(出生時)50,000件中1件 重度の精神発達の遅れ、成長障害、難治性てんかん、多発形態異常。 |
| 5番 | 5p13.2 | コルネリア・デ・ランゲ症候群(Cornelia de Lange症候群) | |
| 5p15.2 | 猫鳴き症候群 | 発生頻度(出生時)20,000〜50,000件中1件 低出生体重、成長障害、甲高い猫のなき声のような啼泣。顔貌所見や筋緊張低下、精神運動発達の遅れ。 5番染色体部分欠失:ねこなき症候群における高解像度マッピング |
|
| 5q35.3 | ソトス症候群(Sotos症候群) | 欠失型と重複型とでは一部症状の差異が指摘されている。 発生頻度(出生時)14,000件中1件 |
|
| 7番 | 7q11.23 | ウィリアムズ症候群(Williams症候群) | |
| 7p13 | パリスター・ホール症候群(Pallister-Hall症候群) | ||
| 7p14.1 | グレイグ脳多合指趾症(Greig 脳多合指趾症) | ||
| 7p21.1 | セートレ・ヒョッツェン症候群(Saethre-Chotzen症候群) | ||
| 7q36.3 | 全前脳胞症3型 | ||
| 8番 | 8q12.2 | チャージ症候群(CHARGE症候群) | |
| 8p23.1 | 8p23.1微細欠失症候群 | ||
| 8q23.3 | 毛髪・鼻・指節症候群1型 | ||
| 8q24.11 | ランガー・ギデオン症候群(Langer-Giedion症候群) | ||
| 11番 | 11p11.2 | ポトツキ・シェファー症候群(Potocki-Shaffer症候群) | |
| 11p13 | WAGR症候群 | ||
| 12番 | 12q24.13 | ヌーナン症候群(Noonan症候群) | RAF1において重複の報告1例、欠失の報告1例 |
| 13番 | 13q14.2 | 網膜芽細胞腫・発達遅滞 | |
| 13q32.3 | 全前脳胞症5型 | ||
| 15番 | 15q11.2〜q13 | プラダー・ウィリー症候群(Prader-Willi症候群) | 父親由来の遺伝子の欠失で、母親由来の遺伝子に起因する 発生頻度(出生時)10,000〜25,000件中1件 筋緊張低下、色素低下、外性器低形成。 |
| 15q11.2〜q13 | アンジェルマン症候群(Angelman症候群) | UBE3Aの機能喪失により発症 発生頻度(出生時)12,000件中1件 重度の精神発達の遅れ、てんかん、失調性運動障害、行動異常、睡眠障害、低色素症、特徴的な顔貌。 |
|
| 16番 | 16p11.2 | 16p11.2微細欠失 | |
| 16p13.11 | 16p13.1微細欠失 | ||
| 16p13.3 | ルビンシュタイン・テイビ症候群(Rubinstein-Taybi症候群) | 発生頻度(出生時)125,000件中1件 | |
| 17番 | 17p13.3 | ミラー・ディカー症候群(Miller-Dieker症候群) | |
| 17p11.2 | スミス・マギニス症候群(Smith-Magenis症候群) | 発生頻度(出生時)15,000~25,000件中1件 | |
| 17q11.2 | 神経芽細胞種1型 | ||
| 20番 | 20p12.23 | アラジール症候群(Alagille症候群) | |
| 22番 | 22q11.2 | ディ・ジョージ症候群(DiGeorge症候群)2型 22q11.2欠失症候群 |
発生頻度(出生時)4,000件中1件 先天性心疾患、精神発達遅延、特徴的顔貌、免疫低下、口蓋裂・軟口蓋閉鎖不全、鼻声、低カルシウム血症。 |
| 22q13.33 | フェラン・マクダーミド症候群(Phelan-McDermid症候群) |
報告例は部分欠失の一部です
東京衛生検査所で使用している次世代シーケンサーは500万塩基以下の欠失・重複は検出できません。
症候群の全ての症例を検出できるわけではありません。
| 染色体 | 重複部 | 症候群 | 備考 |
|---|---|---|---|
| 1番 | 1q21.1 | 1q21.1微細重複症候群 | 1q21.1 部分重複(翻訳) |
| 2番 | 2p21 | 全前脳胞症 | |
| 3番 | 3q29 | 3q29微細重複症候群 | |
| 5番 | 5p13.2 | コルネリア・デ・ランゲ症候群(Cornelia de Lange症候群) | |
| 5q35.3 | ソトス症候群(Sotos症候群) | 欠失型と重複型とでは一部症状の差異が指摘されている。 | |
| 8番 | 8p23.1 | 8p23.1微細重複症候群 | |
| 9番 | 9q34.13 | 結節性硬化症1型 | TSC1遺伝子が原因 5,800件中1人 |
| 10番 | 10q24.3 | 染色体10q24重複症候群 | 11番 | 11p13 | WAGR症候群 |
| 12番 | 12q24.1 | ヌーナン症候群(Noonan症候群) | RAF1において重複の報告1例、欠失の報告1例 |
| 13番 | 13q32.3 | 全前脳胞症5型 | |
| 15番 | 15q26qter | 過成長・知的障碍 | |
| 16番 | 16p11.2 | 16p11.2微細重複 | |
| 16p13.3 | 結節性硬化症2型 | TSC2遺伝子が原因 5,800件中1人 |
|
| 16p13.3 | ルビンシュタイン・テイビ症候群(Rubinstein-Taybi症候群) | CREBBP遺伝子が原因 発生頻度(出生時)125,000件中1件 |
|
| 16p13.11 | 16p13.1微細重複 | ||
| 17番 | 17p11.2 | ポトツキ・ルプスキ症候群(Potocki-Lupski症候群) | 17番染色体(17p11.2)部分重複:発達遅延のある小児における重複 |
| 17p12 | シャルコー・マリー・トゥース病(Charcot-Marie-Tooth)1A型 | ||
| 17q21.31b | 17q21.31微細重複症候群 | ||
| 22番 | 22q11.1 | 猫の目症候群 | |
| 22q11.2 | 22q11.2重複症候群 | 先天性心疾患、精神発達遅延、特徴的顔貌、免疫低下、口蓋裂・軟口蓋閉鎖不全、鼻声、低カルシウム血症。 |
報告例は部分欠失・重複の一部です
東京衛生検査所で使用している次世代シーケンサーは500万塩基以下の欠失・重複は検出できません。
症候群の全ての症例を検出できるわけではありません。
染色体1
部分欠失では、染色体の特定領域に存在する遺伝子が失われ、その結果、発達障害や特定の症候群が引き起こされることがあります。
- 原因: 1番染色体の短腕(1p36)に欠失があることで発生します。
- 症状: 知的障害、発達遅延、筋力低下(低緊張)、行動問題、心臓の異常、顔面の特徴的な異常(例えば、広い前額、浅い目など)が一般的です。その他、けいれん発作や聴覚の問題も伴うことがあります。
1q欠失症候群
- 原因: 1番染色体の長腕(1q)の欠失が原因で発生します。
- 症状: 知的障害や発達遅延、顔の異常、心臓や腎臓の異常が見られることがあります。また、筋力低下やてんかんも報告されています。
1q21.1欠失症候群:1番染色体の長腕(q)の21.1領域が欠失している場合、心臓疾患、精神発達遅延、自閉症スペクトラム障害などのリスクが高まります。
1q42.13欠失症候群(1番染色体の長腕の42.13領域の欠失による症候群)
部分重複では、染色体の特定領域に遺伝子が過剰に存在し、健康や発達に異常が現れます。
1p36重複症候群
- 原因: 1p36領域の遺伝子が重複することにより、遺伝子の過剰発現が引き起こされます。
- 症状: 重複の位置や範囲によって異なりますが、発達遅延、知的障害、特徴的な顔の形態異常が見られます。心臓の異常や筋力低下、行動上の問題(自閉症傾向など)が報告されています。
1q重複症候群
- 原因: 1番染色体の長腕(1q)が重複することで、特定の遺伝子が過剰に存在します。
- 症状: 知的障害、発達遅延、身体的な異常(心臓や腎臓の異常など)や筋力低下が見られることがあります。重複の位置に応じて、症状の重さが異なる場合があります。
染色体2
染色体3
染色体4
4番染色体の部分欠失は、染色体の一部が欠失し、その結果、発達障害や身体的異常が発生することがあります。
4p欠失症候群(Wolf-Hirschhorn症候群)
- 原因: 4番染色体の短腕(4p)の一部が欠失することによって発症します。
- 症状: 重度の知的障害、発達遅延、低身長、特徴的な顔の形態(「兜型の顔」など)、けいれん、心臓の奇形、筋力低下などが見られます。この症候群は珍しい遺伝的疾患として知られています。
4q欠失症候群
- 原因: 4番染色体の長腕(4q)の一部が欠失していることが原因です。
- 症状: 発達遅延、知的障害、心臓や腎臓の異常、顔の形態異常が報告されています。また、筋力低下や運動発達の遅れも見られることがあります。
5番染色体
部分欠失は、染色体の一部が欠失することで、重要な遺伝子が機能しなくなり、健康や発達に悪影響を及ぼすことがあります。
5p欠失症候群(クライ・デュ・シャット症候群)
- 原因: 5番染色体の短腕(5p)の一部が欠失することによって発症します。この症候群は「猫鳴き症候群」とも呼ばれ、特徴的な「猫の鳴き声」に似た泣き声が新生児期に見られることが多いです。
- 症状: 知的障害、発達遅延、筋力低下、特徴的な顔つき(小さな顎、広い鼻、下がった目)、低体重、行動問題が含まれます。重度の症例では、心臓の奇形や呼吸器系の問題が見られることもあります。
5q欠失症候群
- 原因: 5番染色体の長腕(5q)が欠失することで引き起こされる血液疾患です。この異常は骨髄異形成症候群(MDS)と関連し、赤血球を作る細胞に異常が見られることがあります。
- 症状: 貧血、骨髄不全、疲労感、免疫系の異常が報告されています。5q欠失は特に成人女性に多く見られる傾向があります。
6番染色体
7番染色体
7番染色体の部分欠失は、遺伝子の一部が失われることで、健康上の問題を引き起こします。特に以下の症候群がよく知られています。
ウィリアムズ症候群(Williams Syndrome)
- 原因: 7番染色体の長腕(7q11.23)にあるELN(エラスチン)遺伝子の欠失が主な原因です。
- 症状: 知的障害、心臓血管の問題(特に大動脈弁の狭窄)、特徴的な顔つき(小さな鼻、広い口)、社交的な性格が見られます。さらに、発達遅延や学習障害も伴うことがあります。
7q欠失症候群
- 原因: 7番染色体の長腕(q)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、顔面の異常(目の間が広いなど)、成長障害、心臓や腎臓の問題が報告されています。
7番染色体の部分重複は、特定の遺伝子が過剰に存在することで、健康や発達に影響を与えます。以下のような症状が見られる場合があります。
7q11.23重複症候群
- 原因: 7番染色体の同じ領域(7q11.23)が重複することで発症します。ウィリアムズ症候群と逆の状態です。
- 症状: 自閉症スペクトラム障害(ASD)、発達遅延、言語の遅れ、学習障害が主な特徴です。行動問題や感覚過敏も報告されています。
7p重複症候群
- 原因: 7番染色体の短腕(p)が重複することで引き起こされます。
- 症状: 発達遅延、知的障害、特徴的な顔貌、心臓や消化器の問題が見られることがあります。行動障害や睡眠障害も報告されています。
8番染色体:=>ポスター参照
8番染色体の部分欠失は、染色体の一部が失われることにより、発達障害や身体的な異常を引き起こします。以下は、8番染色体の欠失に関連する代表的な症候群です。
8p欠失症候群(8p微細欠失症候群)
- 原因: 8番染色体の短腕(8p)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、特徴的な顔貌、先天性心疾患、運動協調性の問題が見られます。言語発達の遅れや行動上の問題も報告されています。
ラングラー・グルーベル症候群(Langer-Giedion症候群)
- 原因: 8番染色体の長腕(8q)にあるTRPS1遺伝子とEXT1遺伝子の欠失によって引き起こされます。
- 症状: 知的障害、特徴的な顔つき、骨格の異常(特に指や骨の成長障害)、脱毛症(毛髪の成長不全)などが見られます。
9番染色体
9番染色体の部分欠失は、染色体の一部が失われることで発達障害や身体的異常を引き起こします。以下のような症候群が知られています。
9p欠失症候群
- 原因: 9番染色体の短腕(9p)の一部が欠失することにより発症します。
- 症状: 知的障害、発達遅延、特徴的な顔つき(広い鼻、上向きの鼻孔、離れた目など)、成長障害、運動機能の遅れが見られることがあります。心臓や腎臓の異常も報告されています。
- 別名: アルフ・フィース症候群(Alfi’s Syndrome)とも呼ばれます。
9q欠失症候群
- 原因: 9番染色体の長腕(9q)の一部が欠失することで発生します。
- 症状: 知的障害、発達の遅れ、筋力低下、心臓や消化器系の異常、免疫不全などが見られます。欠失した領域に応じて症状の重さが異なります。
10番染色体
10番染色体の部分欠失は、染色体の一部が失われることで、発達障害や身体的な異常を引き起こすことがあります。
10q欠失症候群(10q微細欠失症候群)
- 原因: 10番染色体の長腕(10q)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、特徴的な顔貌、心臓や腎臓の異常が見られることがあります。また、運動機能の遅れや言語の発達遅延も一般的です。
- 治療: 欠失症候群は、早期の介入と療育が重要であり、発達支援や医療的ケアが必要です。心臓の異常などには外科的治療が行われる場合もあります。
10p欠失症候群
- 原因: 10番染色体の短腕(10p)の一部が欠失することで発症します。
- 症状: 発達遅延、知的障害、成長障害、特徴的な顔つき、心臓や腎臓の異常が見られることがあります。顔の特徴的な形態には、広い鼻、上向きの鼻孔、異常に広がった目などが含まれます。
染色体11
11番染色体の部分欠失は、染色体の一部が失われることで、特定の遺伝子の機能が欠如し、発達遅延や身体的な異常を引き起こすことがあります。以下に代表的な症候群を示します。
ウィルムス腫瘍(WT1遺伝子欠失)
- 原因: 11番染色体の短腕(11p)にあるWT1遺伝子の欠失によって発症します。この遺伝子は腎臓の発達に関与し、腫瘍抑制にも重要な役割を果たします。
- 症状: 腎臓のがん(ウィルムス腫瘍)、泌尿生殖器の異常、場合によっては性分化異常を伴います。
ベックウィズ・ウィーデマン症候群(BWS)
- 原因: 11p15.5領域における遺伝子の欠失または機能不全により発症します。これはIGF2などの成長に関与する遺伝子の異常に関連しています。
- 症状: 巨大児症、臓器肥大、へそヘルニア、腫瘍発生のリスクが高いことが特徴です。
ヤコブセン症候群
- 原因: 11番染色体の長腕(11q)の一部が欠失することで発症します。
- 症状: 知的障害、運動遅延、特徴的な顔つき(広い額、下がった目尻)、出血傾向、心臓の異常などが報告されています。
12番染色体
部分欠失は、染色体の特定の領域が失われていることで、さまざまな遺伝子が不足し、その結果として特定の症状が現れる可能性があります。12番染色体における欠失の影響は、欠失した領域とそこに含まれる遺伝子に依存します。
- 症状の例: 知的障害、発達遅延、身体の異常(心臓や腎臓の奇形など)、低身長や筋力低下など。
12q15欠失症候群のように、特定の領域(12番染色体の長腕)が欠失すると、特有の症状が現れることが報告されています。 - 診断: 欠失は染色体検査(FISHやCGHアレイなど)で特定されます。
主な症候群
- 12q欠失症候群:12番染色体の長腕(q)の一部が欠失している場合を指します。一般的な症状には、発達遅延、知的障害、筋緊張低下、特徴的な顔貌、心臓の異常などがあります。
- 12p欠失症候群:12番染色体の短腕(p)の一部が欠失している場合です。これにより、発達遅延や身体的な異常が生じることがあります。

13番染色体
13番染色体の一部が欠失することで、特定の遺伝子が機能しなくなり、発育や健康に影響を及ぼす可能性があります。
- 13q欠失症候群 13番染色体の長腕(q)の部分が欠失することで発症します。具体的な症状には、知的障害、発育遅延、特定の顔貌異常(目が小さい、鼻が低いなど)、指や爪の発達不良、心臓や腎臓の異常が含まれます。
- 網膜芽細胞腫(Retinoblastoma) 13番染色体のq14領域にあるRB1遺伝子の欠失が、網膜芽細胞腫という小児期の眼のがんの原因となることがあります。
13番染色体の部分が重複すると、余分な遺伝情報が含まれるため、過剰な発現が起こり、健康や発達に異常を引き起こす可能性があります。
- 重複症候群: 13番染色体の部分的な重複は非常に稀ですが、知的障害、発育遅延、頭部や顔面の形態異常(例えば、広い額や小さな顎など)が見られることがあります。重複する領域によって症状の重篤度は異なります。
染色体14
14番染色体の一部が欠失すると、その領域に存在する遺伝子が不足するため、発達障害や身体的異常が生じることがあります。
-
症状の例:
- 知的障害や発育遅延
- 特定の顔貌の異常(顔つきの変化や骨格の異常)
- 心臓や腎臓の奇形
- 免疫不全などの症状が報告されています。
- 14q欠失症候群 特に14番染色体の長腕(q)の欠失が知られており、成長障害、筋肉の低緊張、発達の遅れ、さらにはてんかんなどの神経学的症状が伴うことがあります。
染色体15
15番染色体の部分欠失では、以下のような遺伝的疾患が知られています。
プラダー・ウィリー症候群(PWS)
- 原因: 15番染色体の父親由来の一部(通常は15q11-q13領域)の欠失が原因で発生します。まれに遺伝子の機能不全や欠失が原因です。
- 症状: 新生児期からの筋力低下(低緊張)、食欲異常による肥満、知的障害、性腺機能不全などが特徴です。行動上の問題や睡眠障害も見られることがあります。
アンジェルマン症候群(AS)
- 原因: プラダー・ウィリー症候群と同じ領域(15q11-q13)の欠失ですが、こちらは母親由来の染色体で欠失が発生します。
- 症状: 知的障害、運動の協調性欠如、頻繁な笑いや興奮、てんかん発作が主な特徴です。
16番染色体
17番染色体
17番染色体の部分欠失は、染色体の一部が失われることで、重要な遺伝子が不足し、発育や健康に影響を与える可能性があります。
17p13.3欠失症候群(ミラー・ディーカー症候群)
- 原因: 17番染色体の短腕(p)の13.3領域の欠失によって引き起こされます。
- 症状: 重度の知的障害、てんかん、筋肉の低緊張、頭蓋顔面の異常が特徴です。脳の発達に影響を与え、滑脳症と呼ばれる脳の異常を引き起こすことがあります。
スミス・マゲニス症候群(Smith-Magenis syndrome)
- 原因: 17p11.2領域の欠失が原因で発生します。
- 症状: 知的障害、発達遅延、行動異常(例えば、自傷行為)、睡眠障害、特徴的な顔つきなどが見られます。
染色体18
18番染色体の部分欠失は、染色体の一部が欠失することで、遺伝子が失われ、発達や身体の異常が生じます。
18q欠失症候群(18q-症候群)
- 原因: 18番染色体の長腕(q)の一部が欠失することによって発症します。
- 症状: 発育遅延、知的障害、筋力低下、顔面の特徴的な変形、足の変形(扁平足など)、免疫不全、言語の遅れ、行動障害が見られます。また、思春期以降にホルモンの問題が起こることもあります。
18p欠失症候群(18p-症候群)
- 原因: 18番染色体の短腕(p)が欠失することによって発生します。
- 症状: 成長遅延、運動の発達遅延、筋力低下、軽度から中等度の知的障害が一般的です。顔の形態異常、言語発達の遅れなども報告されています。
19番染色体
20番染色体
21番染色体
22番染色体
22番染色体の部分欠失では、遺伝子が欠失することで、発育障害や身体的異常が現れることがあります。特に、22q11.2の欠失が最も一般的です。
キャットアイ症候群(Cat Eye Syndrome)
- 原因: 22番染色体の一部(短腕と長腕の接合部に近い部分)が欠失または重複することで発症します。
- 症状: 目に特徴的な異常(瞳孔が縦に割れる)、肛門や尿路の異常、心臓の異常、発達遅延が報告されています。
2 p deletion
2p25.1–25.3の家族性逆位重複/欠失は、逆位重複の起源に関する新たな手がかりを提供する
私たちは、2p25.3-p25.1 の同じ 10 Mb 逆重複が 2 人の子供とその父親に分離しており、全員がトリソミー表現型を示している家族を研究しました。 FISH 分析により重複が逆転していることが実証されたため、古典的な inv dup del タイプの再配列に従って、連続した末端欠失も存在するのではないかと考えられました。 2p および 2q サブテロメア プローブを使用した FISH では正常な結果が得られましたが、解像度 100 kb のアレイ CGH (aCGH) では、重複の他に 273 kb の欠失も存在することが示されました。正常な対数比を有する有益なスポットは 1 つだけ検出されましたが、高解像度 aCGH 分析 (約 20 kb) によって、欠失領域と重複領域の間に単一コピー領域の存在がさらに疑われました。再配列の正確な構造は、リアルタイム PCR とブレークポイント クローニングによって再定義され、欠失領域と重複領域の間に 2680 bp のシングルコピー配列が存在すること、および非配列を形成する可能性のある単純なリピートが関与していることが実証されました。 B DNA 構造。この再配列は分節重複や短い逆方向反復によって媒介されず、二本鎖切断は非相同末端結合やミクロ相同性媒介鎖内修復によって修復された可能性がある。これらのデータは、逆位重複に関連する付随欠失が、古典的な細胞遺伝学的方法だけで実証できたよりも頻繁に発生する可能性が非常に高いという事実を強調しています。トリソミーおよび末端 2p 欠失の表現型への影響について議論します。
https://www.nature.com/articles/ejhg2008160染色体2p25重複および2q37欠失を有する患者における重度の成長ホルモン欠乏症および下垂体奇形
我々は、重度の成長障害、成長ホルモン(GH)欠乏症、一般的な言語障害を伴う精神運動遅延、および独特の表現型を有する4.8歳で確認された男児について報告する。磁気共鳴画像法(MRI)による視床下部-下垂体領域の評価により、下垂体茎の中断と異所性の下垂体後葉を伴う下垂体形成不全が明らかになり、これらは永久的なGH欠損症の予後マーカーと考えられています。項部の半透明性の増加による出生前の染色体分析では、正常な男性の核型が明らかになりましたが、出生後の高分解能バンドにより 2q 異常の疑いが生じました。その後、アレイ比較ゲノムハイブリダイゼーション (アレイ-CGH) により、2p25 重複と 2q37 欠失からなる de novo 複雑なゲノム再配列が明らかになりました: arr[hg19] 2p25.3p25.1(30,341-9,588,369)x3,2q37.2q37.3( 235,744,424-243,041,305)x1。 FISH分析は、異常な染色体2が、2qの遠位に位置する重複した2p領域を有する逆位の誘導体を模倣していることを示した。これは、我々の知る限り、遠位2p25の重複と2q37の欠失、および下垂体奇形がGH欠損症を引き起こした最初の症例である。
https://molecularcytogenetics.biomedcentral.com/articles/10.1186/1755-8166-7-41肥満、知的障害、および多動性障害の早期発症に関連する1.9 Mbの終末的de novo 2p25.3欠失を有する新規患者
アレイ CGH 解析によって検出された 2p25.3 の末端および間質欠失 (サイズ < Mb) は、知的障害 (ID) および行動上の問題に関連する早発性肥満/過体重に代表される共通の臨床的特徴を共有する約 18 人の患者で報告されています。 。この観察は、2p サブテロメア欠失が症候群性肥満と関連しているはずであり、これまでに発表されたすべての症例で欠失または破壊されているため、MYT1L が ID と肥満の主な候補遺伝子となったという仮説につながりました。
今回我々は、中等度のIDを伴う肥満の早期発症と多動性障害からなる特徴的な表現型を持つ4.4歳の女児において、アレイCGH分析によって検出された、父親由来の1.9Mbの2p25.3のde novo末端欠失について説明した。 。この欠失により MYT1L が破壊され、他の 5 つの OMIM 遺伝子、ACP1、TMEM18、SNTG2、TPO、および PXDN が含まれていました。
ここでは、2p25.5サブテロメア欠失に関連する症候群性肥満の病因において、MYT1Lのヘテロ欠失と一致する可能性のある追加のハプロ不全遺伝子の複合機能効果について議論する。
https://molecularcytogenetics.biomedcentral.com/articles/10.1186/1755-8166-7-532q37欠失症候群:14人の新規患者における過体重、短指症、行動的特徴を含む臨床スペクトルの最新情報
2q37 遺伝子座は、最も一般的に欠失されるサブテロメア領域の 1 つです。このような欠失は、テロメア蛍光 in situ ハイブリダイゼーション (FISH) 分析によって 100 名を超える患者で確認されており、頻度は低いですが、アレイベースの比較ゲノム ハイブリダイゼーション (アレイ CGH) によっても確認されています。認識可能な「2q37欠失症候群」またはオルブライトの遺伝性骨異栄養症様症候群は、以前に報告されています。欠失をより適切にマッピングし、この欠失症候群をさらに改良するために、我々はフランス語細胞遺伝学者協会と協力して、FISH およびアレイ CGH によって特徴付けられる遠位または間質性 2q37 欠失を持つ新たな知的障害患者 14 人を収集しました。患者は顔面異形症(13/14人)と短指症(10/14人)を示し、これは行動上の問題、自閉症またはさまざまな重症度の自閉症スペクトラム障害、および過体重または肥満に関連していた。これら 14 人の新規患者の欠失は 2.6 ~ 8.8 Mb でした。 HDAC4 の主要な役割は証明されていますが、欠失領域における他のいくつかの遺伝子の表現型の関与は不明です。我々は、2q37 欠失の遺伝子型と表現型の相関をさらに精密化しました。これを行うために、臨床データ、文献レビュー、および Manteia データベースを使用して、骨格奇形 (顔面異形症および短指症)、過体重、行動上の問題および発作の候補遺伝子の最小の重複欠失領域を調べました。同定された候補遺伝子のうち、PRLH、PER2、TWIST2、CAPN10、KIF1A、FARP2、D2HGDH、PDCD1の役割に焦点を当てます。
https://www.nature.com/articles/ejhg20122302p25.1–25.3の家族性逆位重複/欠失は、逆位重複の起源に関する新たな手がかりを提供する
私たちは、2p25.3-p25.1 の同じ 10 Mb 逆重複が 2 人の子供とその父親に分離しており、全員がトリソミー表現型を示している家族を研究しました。 FISH 分析により重複が逆転していることが実証されたため、古典的な inv dup del タイプの再配列に従って、連続した末端欠失も存在するのではないかと考えられました。 2p および 2q サブテロメア プローブを使用した FISH では正常な結果が得られましたが、解像度 100 kb のアレイ CGH (aCGH) では、重複の他に 273 kb の欠失も存在することが示されました。正常な対数比を有する有益なスポットは 1 つだけ検出されましたが、高解像度 aCGH 分析 (約 20 kb) によって、欠失領域と重複領域の間に単一コピー領域の存在がさらに疑われました。再配列の正確な構造は、リアルタイム PCR とブレークポイント クローニングによって再定義され、欠失領域と重複領域の間に 2680 bp のシングルコピー配列が存在すること、および非配列を形成する可能性のある単純なリピートが関与していることが実証されました。 B DNA 構造。この再配列は分節重複や短い逆方向反復によって媒介されず、二本鎖切断は非相同末端結合やミクロ相同性媒介鎖内修復によって修復された可能性がある。これらのデータは、逆位重複に関連する付随欠失が、古典的な細胞遺伝学的方法だけで実証できたよりも頻繁に発生する可能性が非常に高いという事実を強調しています。トリソミーおよび末端 2p 欠失の表現型への影響について議論します。
https://www.nature.com/articles/ejhg2008160染色体 2q 末端欠失: 6 人の新規患者の報告と 66 人における表現型とブレークポイントの相関関係のレビュー
我々は、2q36 に切断点を持つ 2 番染色体の末端欠失を有する新規患者と、2q37 に切断点を有する 2q 末端欠失を有するさらに 5 人の新規患者を報告する。片側横隔膜ヘルニアは、2q37.1 にブレークポイントを持つ 1 人の患者における新規所見です。これらの患者を、以前に報告された2q末端欠失を持つ60人の患者と比較すると、特定の身体的異常はブレークポイントの位置と大まかに関連していることがわかりました。例えば、2q37.3 またはその近位にブレークポイントがある患者では、顔の特徴 (例: 突出した額、下がった鼻梁、異形の耳と鼻)、低身長、短い手と足が頻繁に見られました。馬蹄形腎臓およびウィルムス腫瘍の報告は、2q37.1 にブレークポイントがある患者に限定されており、構造的脳異常および気管異常は 2q37.1 またはその近傍にブレークポイントがある患者でのみ報告されていました。口蓋裂は、最も近位のブレイクポイント (2q36 または 2q35) を持つ患者でのみ報告されました。この患者集団では、発達遅延、精神遅滞、自閉症様行動、筋緊張低下などの神経学的影響が典型的でしたが、ブレークポイントによる重症度の階層化はありませんでした。顕著な筋緊張低下、哺乳不良、胃食道逆流、発育遅延のある乳児、発達遅延、自閉症行動、本明細書に記載の特徴的な顔面および外皮の特徴のある年長児では、第2染色体長腕の末端欠失を考慮する必要がある。臨床的特徴を特定のブレークポイントに割り当て、予測値を改良することは、カウンセリングに役立つ可能性があります。
https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.301562q37.3 de novo サブテロメア欠失のアレイ CGH 解析と臨床的説明
私たちは、正常核型を持ち、サブテロメア FISH 分析によって検出された、染色体 2q の新規の潜在的な末端欠失を持つ 13 歳の少女について報告します。 1 Mb 分解能の Spectral Chip 2600 (Spectral Genomics) を使用したアレイ CGH 分析によるさらなる調査により、欠失が確認され、さらに 4 つの追加クローンの欠失も示されました。アレイ CGH では他の異常は検出されませんでした。欠失の詳細なマッピングのために 8 つの BAC プローブを使用した FISH 研究が実行され、アレイの結果が確認されました。 FISH分析により、欠失ブレークポイントはクローンRP11-84G18とRP11-83N2の間にあり(クローン間の物理的距離は0.36Mb)、テロメアにまで及んでいることが示された。欠失のサイズは約 6.4 ~ 6.7 Mb と推定されました。臨床所見には、発達遅延、重度の行動障害、成長思春期遅滞、先天性軽度伝音難聴、成長ホルモン欠乏症、代償性甲状腺機能低下症、異形の顔貌、過度の関節可動性亢進、短中指骨、異常な皮膚痕跡、および新生児喉頭軟化症、筋緊張低下、皮膚痕跡の病歴が含まれます。臍ヘルニア。私たちの患者の表現型は、観察されなかった心血管、泌尿生殖器、神経学的異常および湿疹を除いて、文献の表現型と一致しています。同様の症例の臨床的および分子的症状の報告により、表現型と遺伝子型の正確な相関関係と、家族の適切な遺伝カウンセリングが可能になります。
https://www.sciencedirect.com/science/article/abs/pii/S1769721206001005?via%3Dihubアレイ比較ゲノムハイブリダイゼーションを使用して検出された滑脳症および 2p25.3 および 2q37.3 微小欠失を伴うリング 2 番染色体の出生前診断
我々は、IUGR、小頭症、滑脳症、および不明瞭な外生殖器を有する胎児の未培養羊膜細胞を使用した、aCGHによる2p25.3および2q37.3微小欠失を有する環状染色体2の異数性迅速診断を提示する。私たちの症例では、2p25.3 および 2q37.3 の微小欠失を伴う、環染色体 2 の CNS 異常のリストに滑脳症が追加されました。この場合、2q37.3 における HDAC4、KIF1A、PASK、HDLBP、FRAP2、および D2HGDH のハプロ不全、および 2p25.3 における MYT1L、SNTG2、および TPO のハプロ不全の影響について議論します。
https://www.sciencedirect.com/science/article/abs/pii/S0378111913001285?via%3Dihubコロンビアの大頭症患者における2q37欠失症候群:症例報告
背景
2q37欠失症候群は、2q37サイトバンドの欠失によって引き起こされる稀な常染色体優性疾患で、発達遅延、知的障害、行動異常、頭蓋顔面の異形を引き起こし、世界中で115人以上の患者が報告されています。
事例紹介
言語コミュニケーションの遅れ、臍ヘルニア、顔面異形、筋緊張低下、および大頭症を患うコロンビア人の 3 歳の患者について、正常な磁気共鳴画像法で報告します。マイクロアレイベースの比較ゲノムハイブリダイゼーションにより、2q37.2および2q37.3領域に5.9Mbの欠失が明らかになり、第2染色体の1つにある60個のタンパク質コード遺伝子が除去され、この患者の2q37欠失症候群の診断が可能になった。これまでの治療介入は臍ヘルニアの外科的矯正でした。
結論
遺伝子検査は、臨床的に複雑で頻度の低い症状を診断するためだけでなく、適切な監視、介入、遺伝カウンセリングを可能にするタイムリーな診断にも重要なツールです。この症例は、2q37 欠失症候群の表現型および遺伝的特徴付けを拡張するための情報も提供します。
3 p deletion
知的障害と異形の特徴を持つ患者における3p25.3の欠失と、重要領域のさらなる定義
最近、3番染色体短腕末端の間質性欠失に関するいくつかの報告がなされ、その欠失が3p欠失症候群の原因となる重要な領域の特定に役立っている。われわれは、知的障害、強迫傾向、筋緊張低下、異形顔貌を有する11歳の女児で、アレイCGHを用いて684kbの間質性3p25.3欠失が特徴付けられた症例について報告する。この欠失は、最近報告された3例の間質性3p25欠失と重複する。これらの欠失は、THUMPD3、SETD5、LOC440944の3つのRefSeq注釈付き遺伝子のみを含む124kbの重複領域を共有していた。本症例は、知的障害、筋緊張低下、鼻梁陥凹、長い顎堤などの表現型が、これまでに報告された症例と類似していたが、より大きな欠失を有する症例で報告されている心臓障害、発作、小頭症は認めなかった。したがって、この患者は、遺伝子型と表現型の相関を示唆すると同時に、3p欠失の結果に関するわれわれの知識をさらに深めるものである。
https://pubmed.ncbi.nlm.nih.gov/23613140/遠位3p欠失症候群:3つの小さな遠位欠失の詳細な分子細胞遺伝学的および臨床的特徴と総説
遠位型3p欠失症候群は、発達遅延、低出生体重、発育遅延、小頭症、腕頭症、眼瞼下垂、長い口唇、小顎症、低置耳を特徴とする。我々はFISHとBACを用いて、3つの3p欠失を分子レベルで詳細にマッピングした。欠失の大きさは10.2-11 Mbで、VHL遺伝子を含む47-51の既知の遺伝子を包含していた。欠失の1つは間質性で、3pテロメアは無傷であった。過去に発表された3p欠失を有する9例の患者において、分子細胞遺伝学的または分子細胞遺伝学的手法を用いて欠失の大きさが推定された。これらの症例の注目遺伝子を含む遺伝子型と表現型を比較し、考察した。われわれの患者の1人における近位ブレイクポイントの局在は、以前に同定された心臓欠損の臨界領域が、現在3つの候補遺伝子を含んで絞り込まれている可能性を示唆している。また、ATP2B2遺伝子の欠失だけでは、3p欠失の患者にしばしば認められる聴覚障害を引き起こすには不十分であると結論できる。これは、遠位3pの間質性欠失で報告された3例目の症例である。
https://pubmed.ncbi.nlm.nih.gov/17696125/3p欠失症候群の特徴を有する患者における3p25上の微小欠失
稀な3p欠失症候群は、3番染色体短腕の様々な長さの欠失によって引き起こされる異常のスペクトルを示す。これらの欠失の多くは3p末端を含むが、間質性欠失も本症候群の特徴を生じることがある。我々は、典型的な3p欠失の特徴を多く示す患者において643kbの間質性欠失を検出した。この患者は、以前に報告された1.6Mbの間質性欠失を有する患者と共通する多くの所見を有しており、その中には認知障害、発作、先天性心欠損などが含まれていた。12の遺伝子を含む518kbの重複領域が、これらの特徴のいくつかにとって重要な領域であることが証明された。CRELD1、SRGAP3、CAMK1、TADA3、MTMR14などのいくつかの遺伝子の推定される機能について、3p欠失症候群の表現型に関与する可能性に関して考察した。我々は、この518kbの重複領域が、欠失した場合に3p欠失症候群の表現型を生じさせる重要な領域を規定する可能性があることを示唆している。
https://pubmed.ncbi.nlm.nih.gov/22903836/2家系における末端3p欠失–分子核型と表現型の相関性
3p欠失症候群は、3p25-pter領域の異なるサイズの欠失によって引き起こされるまれな疾患である。成長遅延、発達遅延、精神遅滞、異形、小頭症、眼瞼下垂を特徴とする。欠失を有する個体の表現型は正常から重度まで様々である。ほとんどの症例はde novoで起こるが、少数の家族性症例が報告されている。我々は、末端3p欠失を有し、臨床的特徴が極めて多様な2つの家族について記述する。A家系では、母親と娘は極めて軽症であったが、息子はより重症であった。B家系では、母親は正常で、息子は罹患しており、これまでの3p欠失症候群では報告されていない症状を呈していた。欠失はゲノムワイドSNPアレイ解析によって特徴づけられ、大きさは9Mbと1.1Mbであった。
CHL1、CNTN4、CRBN遺伝子の塩基配列解析では、プロバンドのより重篤な表現型を説明する可能性のあるマスクされた劣性対立遺伝子は発見されなかった。A家系では、9 Mbの欠失がプロバンドの3p欠失症候群の原因と考えられるが、他の家系の極めて軽度の表現型は依然として説明できない。B家系では、1.1 Mbの末端欠失はCHL1遺伝子のみを包含しており、3p欠失症状を引き起こすには不十分である。染色体および/またはゲノムワイド・アレイの分析所見に基づいて子孫の表現型を予測することができないためである。
3p25.3バンドの欠損はdel(3p)症候群の発現に重要である:3番染色体短腕遠位部の欠損症例における核型-表現型の相関性
3番染色体短腕遠位部のモノソミー患者2例について述べる。一人は46,XX,del(3)(p25.3)、もう一人は46,XX,r(3)(p26.1q29)であった。前者の患者には、成長障害、精神遅滞、扁平後頭部を伴う小頭症、三角顔、合瞼症、眼瞼下垂症、過眼瞼症、広くて平坦な鼻、長い顎堤、下を向いた口、小顎症、明らかに低く奇形な耳、指の異常、難聴などの3p-症候群の特徴的な臨床症状がみられた。後者の患者は、精神遅滞、成長不全、小頭症を伴う非特異的な表現型であった。本症例と、これまでに報告された3pの遠位部分の欠損を伴う16症例の核型-表現型の比較から、3p25.3バンドの欠損がdel(3p)症候群の主な臨床症状を引き起こすのに重要であることが示唆された。
https://pubmed.ncbi.nlm.nih.gov/2178418/非平衡転座に起因する3p25.3→pterモノソミーと結合した部分的モノソミー21(q11.2→q21.3):形態異常と発達遅滞を呈した1例
神経精神運動発達遅滞、聴覚障害、形態異常のため遺伝学的評価のために紹介された1歳5ヵ月の女性患者について述べる。患者は、アンバランスなde novo転座による21番染色体の部分モノソミー(q11.2→q21.3)と3p染色体の末端モノソミー(p25.3→pter)を合併していた。転座は蛍光in situハイブリダイゼーション(FISH)で確認され、ブレークポイントは高分解能アレイでマッピングされた。これらの技術を組み合わせた解析の結果、最終的な核型は45,XX,der(3)t(3;21)(p25.3;q21.3)dn,-21.ish der(3)t(3;21)(RP11-329A2-,RP11-439F4-,RP11-95E11-,CTB-63H24 +)と定義された。 マイクロサテライトDNAマーカーの解析から、染色体再配列の起源は父系であることが示唆された。これは、de novoの不均衡転座による3p末端モノソミーと結合した部分的な近位モノソミー21を有する初めての症例である。
https://www.sciencedirect.com/science/article/pii/S03781119120107483p-症候群は3p25.3に難聴遺伝子座を定義する
染色体3pの末端に影響を及ぼす欠失は、3p-症候群と呼ばれる一連の特徴的な臨床症状をもたらす。両側の感音性難聴(SNHL)は、すべての症例ではないが、いくつかの症例で認められ、3p25バンドにある重要な遺伝子の欠損による可能性が示唆されている。現在までのところ、この領域にヒトの難聴を引き起こす遺伝子座は示されていない。しかし、ATP2B2遺伝子は3p25.3に位置しており、マウスのホモログのハプロ不全は同様の重症度のSNHLをもたらす。われわれは、これまでに報告されていない7例の3p-症候群患者の聴覚検査結果と微細な欠失マッピングを比較し、欠失が中等度から重度の両側性SNHLに関連する3p25.3の1.38 Mb領域を同定した。この新規難聴遺伝子座には、ATP2B2を含む18の遺伝子が含まれている。ATP2B2は細胞膜カルシウムポンプPMCA2をコードしている。ヒト蝸牛切片の免疫組織化学的解析から、PMCA2が有毛細胞の定位繊毛に存在することが示され、聴覚系におけるその機能がヒトとマウスの間で保存されていることが示唆された。この領域の他の遺伝子も候補として残っているが、我々は、ATP2B2のハプロ不全が3p-症候群におけるSNHLの最も可能性の高い原因であると結論づけた。
https://www.sciencedirect.com/science/article/abs/pii/S0378595506003145間質性近位3p欠失: 臨床的に認められる症候群
体質異常として生じる3番染色体近位短腕の間質性欠失はまれであり、定義された臨床表現型はまだ確立されていない。我々は、3p12染色体の間質性欠失(del(3)(p12p12))に伴う明瞭な顔貌(方形顔貌、斜頭、広い額、広い鼻梁、長い顎堤、低い耳)を有し、精神運動/言語遅滞を呈する生後30ヵ月の女児について報告する。近位3p欠失症候群をさらに明確にするために、本児の臨床症状を、以前に報告された同じ欠失を有する他の8例の患者と比較した。
https://www.sciencedirect.com/science/article/abs/pii/S038776040600235X発達遅延と先天性心欠損を有する患者における染色体r(3)(p25.3q29): 症例報告と文献的考察
環状3番染色体(r(3))は極めて稀な細胞遺伝学的異常であり、臨床的に不均一であり、文献的には12例しか報告されていない。ここでは、r(3)を有し、42の既知の遺伝子を含む染色体3pterp25.3(61,891-9,979,408)の約10-MBの欠失がG-バンディング核型検査とCytoScan 750K-Arrayを用いて検出された、特徴的な顔貌、発達遅滞、先天性心疾患を呈する1歳の女児について報告する。r(3)のブレイクポイントは3p25.3と3q29にマッピングされた。我々はまた、遺伝子型と表現型の相関に関するより貴重な情報を提供するために、報告されたr(3)および3p欠失症候群の症例の臨床的特徴に関する利用可能な情報を分析した。我々の知る限り、これはr(3)症例で報告された最大の検出断片であり、全ゲノムマイクロアレイを用いた2番目のr(3)研究である。
https://karger.com/cgr/article-abstract/148/1/6/62466/Chromosome-r-3-p25-3q29-in-a-Patient-with?redirectedFrom=fulltext3p欠失症候群患者における微妙な間質性del(3)(p25.3p26.2)の分子細胞遺伝学的解析
3p欠失症候群は、特徴的な顔貌、成長障害、精神遅滞を伴う。通常、3p欠失症候群の個体は、3p25から3pterまでの物質が欠損する末端欠失を有する。今回われわれは、3p欠失症候群に一致する臨床表現型(眼瞼下垂、小頭症、成長障害、発達遅延)を有し、3番染色体短腕の遠位部に微妙な間質性欠失を有する小児を紹介する。3pサブテロメリックプローブを用いた蛍光in situハイブリダイゼーション(FISH)法により、3番染色体の末端領域が存在することが確認された。染色体領域3p25-p26にマッピングされたSequence Tagged Sites(STS)連結BACクローンを用いて、FISHによる間質性欠失の特徴を明らかにした。その結果、欠失はSTSマーカーD3S3630とD3S1304の間の約4.5Mbの領域内にあることが示された。この間質性欠失は、これまでに報告された欠失性3p症候群の末端欠失の全てに含まれ、欠失性3p症候群に関連して報告された最小の欠失である。この欠失の特徴を明らかにすることは、欠失3p症候群の表現型にヘミ接合体で存在する場合に寄与する、成長と発達に重要な遺伝子を同定するのに役立つかもしれない。
https://onlinelibrary.wiley.com/doi/10.1002/ajmg.10323染色体4p16と3p26.3の再配列と多様な臨床像を示すインドの大家族
背景
染色体4p16.3のWolf-Hirschhorn症候群臨界領域(WHSCR-2)の欠失は、典型的には、特徴的な顔貌、様々な知的障害、定型、出生前の成長遅滞の発症をもたらすが、同じ染色体領域の利得は、より様々な程度の知的障害と異形性をもたらす。同様に、3p染色体の末端欠失(3p欠失症候群)の表現型は、軽度から重度の知的障害、小頭症、三頭症、明瞭な顔貌まで様々である。
方法と結果
染色体マイクロアレイ解析と蛍光in situハイブリダイゼーション解析により、染色体サブ領域4p16.1と3p26.3を含む複雑な再配列が明らかになり、3人に4p16.1欠失と3p26.3重複、7人に4p16.1重複と3p26.3重複が認められた。4p16.1欠失と3p26.3微小重複を有する3例全てにWHSの典型的な臨床症状が認められた。4p16.1重複と3p26.3微小重複を有する症例では、典型的な3p微小欠失や4p部分トリソミー症候群から、明瞭な形態異常を伴うより重篤な神経発達遅滞まで、様々な臨床症状が認められた。
結論
我々は、複雑なt(4p;3p)染色体再配列を有し、罹患者にWolf Hirschorn症候群、3p欠失症候群、4p重複症候群を含む多様な臨床転帰を示す最大規模の血統を提示する。
8 p deletions / duplications
8p23.1 partial deletion
モノソミー8pは、8染色体の一部の欠失を特徴とするまれな染色体異常である。 8p23.1欠失の発生率は、羊水サンプルで18542人に1人、出生後サンプルで5072人に1人と推定された。
https://pubmed.ncbi.nlm.nih.gov/10521848/8p23.1 partial duplication
8p23.1重複症候群(8p23.1 DS)は発生頻度が高く、推定有病率は58,000人に1人である。
https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.371209 p deletions
染色体9pの遠位欠失を有する10人の患者の詳細な特徴付けと臨床的相関性
9p遠位端の欠失は、三頭脳、精神遅滞、異形顔貌、心臓異常、生殖器異常と関連している。これまでの研究で、9pテロメアから11.8Mbと16Mbの間にあるバンド9p23に、コンセンサス表現型に重要な領域があることが示唆されている。9人は9p-症候群と一致する臨床的特徴を有するが、多くの報告例より小さい末端欠失を有する。一方、1人は9p-表現型を欠き、母親から受け継いだ140kbの間質性テロメア欠失を示す。
蛍光in situハイブリダイゼーションとマイクロアレイ解析を組み合わせて、それぞれの欠失の大きさを明らかにした。
結果
欠失の大きさは、臨床的に関連する表現型を持つ患者で800kbから12.4Mbまで様々であった。臨床的評価と比較では、欠失の大きさによる身体的特徴の差はほとんど認められなかった。臨床的に関連する表現型を持つすべての患者において、重度の言語障害が観察された。
結論
9p-に一致する表現型を示す我々の患者に共通する最小の欠失領域は、6つの既知の遺伝子を含む9pterの2Mb未満である。これらの遺伝子は9p欠失症候群の主要な特徴のいくつかに寄与している可能性がある。
家族性バランス転座により 9p症候群の表現型を示す子孫 9p症候群
我々は、一卵性双生児姉妹の子供である2人の同様の罹患したいとこについて報告する。 一卵性双生児姉妹の子供)である。9p欠失症候群と一致する表現型の特徴を有する2人のいとこ(一卵性双生児の姉妹の子供)について報告する。(三頭筋頭蓋、中顔面低形成、上方傾斜口蓋裂、長い口唇など知的障害、性発達障害などである。 性発達障害。最初の細胞遺伝学的検査では プロバンドとその両親の核型は正常であった。 マルチプレックス・ライゲーション・プローブ増幅法により、1q末端重複と9p末端欠失が明らかになった。重複と9p末端欠失が認められた。 蛍光in situハイブリダイゼーションによるさらなる解析の結果、以下のことが同定された。 家族性のバランスのとれたクリプティック転座t(1;9)(q44;p23)が母親において同定された、 臨床遺伝学における分子細胞遺伝学的手法の重要性を示すものである。
https://www.funpecrp.com.br/gmr/year2014/vol13-2/pdf/gmr3782.pdf染色体9pの遠位欠失を有する10人の患者の詳細な特徴と臨床的相関
目的
9p遠位端の欠失は、三頭脳、精神遅滞、異形顔貌、心臓異常、生殖器異常と関連している。これまでの研究で、9pテロメアから11.8Mbと16Mbの間にあるバンド9p23に、コンセンサス表現型に重要な領域があることが示唆されている。9人は9p-症候群と一致する臨床的特徴を有するが、多くの報告例より小さい末端欠失を有する。一方、1人は9p-表現型を欠き、母親から受け継いだ140kbの間質性テロメア欠失を示す。
方法
蛍光in situハイブリダイゼーション(FISH)とマイクロアレイ解析を組み合わせて、それぞれの欠失の大きさを明らかにした。
結果
欠失の大きさは、臨床的に関連する表現型を持つ患者において、800kbから12.4Mbまで様々であった。臨床的評価および比較では、欠失の大きさによる身体的特徴の差はほとんど認められなかった。臨床的に関連する表現型を持つすべての患者において、重度の言語障害が観察された。
結論
9p-に一致する表現型を示す我々の患者に共通する最小の欠失領域は、6つの既知の遺伝子を含む9pterの2Mb未満である。これらの遺伝子は9p欠失症候群の主要な特徴のいくつかに寄与している可能性がある。
9p染色体の末端9Mb欠失患者: 三頭脳を伴う9pモノソミー症候群の重要領域の絞り込み
9pモノソミー症候群の典型的な症状である三頭筋頭蓋と性転換を有する患者について述べる。アレイ比較ゲノムハイブリダイゼーション(CGH)により、9p23を切断点とする約9Mbの9p末端欠失が明らかになった。われわれは、三頭頭症を伴う9pモノソミー症候群の報告例と関連する9pの欠失セグメントを比較した。しかし、他の染色体の材料を含まない純粋な末端欠失または間質欠失のみを比較したところ、D9S912からRP11-439I6までの約1Mbの領域がすべての患者で欠失されていることが確認された。我々は、この1Mbの領域が、三頭脳を伴う9pモノソミー症候群の重要な領域である可能性を提唱する。
https://onlinelibrary.wiley.com/doi/10.1111/j.1741-4520.2012.00362.x9p欠失症候群のオランダ人患者13人の臨床的および細胞遺伝学的特徴づけ: コンセンサス表現型に重要な領域の特定
9p欠失症候群は、9番染色体短腕の一部の体質的モノソミーによって引き起こされる。臨床的には、異形顔貌(三頭症、中顔面低形成、長い顎堤)、筋緊張低下、精神遅滞を特徴とする。9p欠失は不均一であることが知られており、様々な大きさの欠失を示す。コンセンサス表現型にとって重要な領域は、9p22上の約4-6Mbの区間に位置することが報告されている。本研究では、13例のオランダ人患者を対象に、蛍光in situハイブリダイゼーション(FISH)と、一部の症例ではアレイベースの比較ゲノムハイブリダイゼーション(アレイCGH)により欠失ブレイクポイントを決定した。様々な発生学的特徴について、遺伝子型と表現型の明確な相関は確立できなかった。しかし、9p欠失症候群の重要な領域を約300kbに絞り込むことができた。三頭症の機能的候補遺伝子であるCER1遺伝子は、この領域のすぐ外側に位置しているようであった。さらに9人の孤立性三頭筋頭蓋症患者のこの遺伝子の塩基配列解析では、病因となる突然変異は発見されなかった。
https://onlinelibrary.wiley.com/doi/10.1002/ajmg.a.32310ミラーハンドの新たな候補領域:末端9p欠失と20p重複を有する2人の患者
9p欠失症候群は、これまでの研究で詳細に定義されており、精神運動発達遅滞、異形、生殖器異常などさまざまな臨床的特徴が特徴であった。9p欠失症候群とは対照的に、20p重複は文献上ほとんど報告されておらず、症例報告が数例あるのみであった。9p欠失症候群と20p重複の合併については、わずか4例しか報告されていない。本研究では、鏡視下手術を有する2名の患者に観察された、9p部分モノソミーと20pトリソミーという稀な染色体再配列を調査することを目的とした。ミラーハンド運動は、遺伝的要因と環境的要因の組み合わせによって影響を受けていた。DCC、NTN1、RAD51、DNAL4の変異に関連する症例もあるが、鏡手運動の遺伝的基盤が説明できない症例も多かった。我々の患者では、これまでミラーハンドの原因として知られていた遺伝子には変化が検出されなかった。この新たな発見は、9p欠失領域または20p重複領域内の遺伝子の用量効果、あるいはブレークポイント領域内で破壊された遺伝子に起因する可能性がある。今後、この遺伝子座内の遺伝子に焦点を当てた研究が進めば、ミラーハンド運動の新たな病因を発見できる可能性がある。
https://link.springer.com/article/10.1007/s11033-023-09192-99p欠失症候群の心臓表現型に関する洞察: イタリアの多施設における経験と文献レビュー
染色体9p欠失症候群は、先天性心疾患(CHD)を含む広範な臨床的特徴を呈するまれな常染色体優性疾患である。現在までのところ、この疾患に関連する心臓の表現型と機能の深い特徴付けに焦点を当てた研究は不足している。われわれは、9p欠失症候群と分子診断された患者10人のコホートについて、多施設共同前向き観察研究を実施し、従来の心エコー法と組織ドップラー画像エコー法を用いて完全な心臓学的評価を行った。その結果、主要なCHDを持たない9p欠失症候群患者が、不顕性心構造変化と左室収縮期および拡張期機能障害を示す可能性があることを示すことができた。より大規模なコホートでの検証が必要であるとはいえ、われわれの所見は、9p欠失症候群患者において完全な心臓評価を行い、長期的な経過観察に組み込むべきであるという考えを支持するものである。
https://www.mdpi.com/2073-4425/14/1/146自閉症と精神遅滞を伴う女児における9pの欠失と重複を伴う新規染色体転座t(11;9)(p15;p23)
自閉症と精神遅滞を呈した5歳の女児について述べる。従来の核型分析により、新規の一方向転座t(11;9)(p15;p23)が明らかになった。HumanCytoSNP-12チップ解析により、9p24.3から9p23までの13Mbの欠失と、9p23から9p21.2までの12.5Mbの重複が同定された。核型は45,XX,psu dic(11; 9)(p15;p23)と記載され、今回初めて報告された。9p24.3から9p23に及ぶ欠失領域は、モノソミー9p症候群の候補領域と重なり、自閉症スペクトラム障害(ASD)の可能性のある遺伝子座を含んでいる。9p23から9p21.2に伸びる重複領域は、9p重複症候群の重要な領域として以前に同定されていた。これらの結果は、一見バランスのとれたde novo転座が隠微な欠失や重複を生じる可能性を示唆しており、異常領域の正確なマッピングは臨床管理を改善する可能性がある。
https://www.sciencedirect.com/science/article/abs/pii/S037811191200462318 p deletions
汎下垂体炎を呈した(新生)変異
18番染色体短腕の欠失は、知的障害、成長遅延、頭蓋顔面奇形(突出耳、小頭症、眼瞼下垂、丸顔など)を特徴とするまれな疾患である。その表現型スペクトラムは広く、軽度の先天奇形から全頭症までさまざまな異常が含まれる。われわれは、眼瞼下垂、丸顔、後頭部の髪の生え際が低い広頚、低身長、汎下垂体症を有する2歳の女児の症例を紹介する。中耳炎再発のため人工呼吸チューブ挿入術を受けた。脳磁気共鳴画像では、異所性下垂体後葉、および下垂体茎の可視化が乏しく浅く小さいトルコ鞍が示された。細胞遺伝学的解析および染色体マイクロアレイ解析により、18番染色体の短腕にde novo欠失が認められた(arr 18p11.32p11.21[136,227-15,099,116]x1)。GH欠乏症と診断され、生後6ヵ月より遺伝子組換えヒト成長ホルモン(GH)療法を受けている。彼女の成長率は、GH治療による副作用なしに改善した。この症例は、18p欠失症候群の表現型スペクトラムを拡大し、成長不全を特徴とするこの症候群において、GH治療が直線的成長にプラスの影響を与えることを強調している。18p欠失症候群における欠失の大きさや遺伝子座に応じた遺伝子型と表現型の相関を明らかにし、予後を予測するためには、さらなる研究が必要である
https://pubmed.ncbi.nlm.nih.gov/30943682/18p染色体欠失を有する成人男性の追跡調査
18p-症候群は40年以上前から知られており、最初の報告はde Grouchyらによるものであった[Comptes Rendus Hebdomadaires Séances l’Acad Sci 256 (1963) 1028]。様々な重症度の精神遅滞が最も不変の特徴である。 100例以上の症例が報告されている。最も高齢の患者は50歳である [Hum Genet 63 (1983) 139; Clin Genet 2 (1971) 338]。当時22歳と42歳であった2人の成人患者[Ann Génét 29 (1986) 107]の追跡調査が報告されており、現在42歳と62歳である。18p-症候群の成人患者の経過をより明確にするためには、さらなる症例報告が必要である。
https://www.sciencedirect.com/science/article/abs/pii/S1769721205000303?via%3Dihubターナー症候群に類似した低身長、短い頚部、低い後頭部のヘアライン、腫れぼったいまぶた、肘関節の屈曲角増加などの臨床的特徴を呈した18p欠失症候群の13歳女児
Turner症候群に類似した臨床症状を呈する18p欠失症候群の13歳女児を報告する。
症例報告
13歳の女児が、低身長、短網頸、低い後頭髪線、腫れぼったいまぶた、肘の携帯角増大というターナー症候群様の臨床的特徴を呈し、遺伝カウンセリングのために紹介された。女児はまた、軽度の知的障害、精神運動発達遅滞、言語障害、高円唇口蓋、多指症、中顔面低形成を有していた。この女児の細胞遺伝学的解析の結果、核型は46,XX,del(18)(p11.2)であった。両親の核型は正常であった。末梢血から抽出したDNAのアレイ比較ゲノムハイブリダイゼーション解析の結果、18p11.32-p11.21またはarr 18p11.32p11.21の13.93-MB欠失が明らかになった。 21(148,993-14,081,858)×1.0[GRCh37(hg19)]が欠失し、USP14、TYMS、SMCHD1、TGIF1、LAMA1、TWSG1、GNALおよびPTPN2を含む52のOnline Mendelian Inheritance in Man(OMIM)遺伝子が含まれていた。多型DNAマーカー解析により、欠失の起源は母体由来であることが明らかになった。
結論
知的障害、顔面異形、精神運動発達遅滞を伴うターナー症候群様の臨床的特徴を有する女性は、染色体欠失症候群を疑うべきである。
染色体18p欠失症候群と非対称性中隔肥大症を合併した乳児の1例
18p欠失症候群の頻度は5万人に1人程度と推定され、低身長、知的障害、顔面異形などの臨床的特徴を伴うことが多い。本症例の身体所見では、低身長、知的障害、顔面異形(小頭症、眼瞼下垂、外反母趾、低い鼻梁、突出した耳、長い指頭、薄い唇)、および第5指の指切断が認められた。周辺核型は46、XX、del(18)(p11.32p11.2)であった。DNAマイクロアレイ解析の結果、18p11.32p.11.21に13.9-MBのde novo欠失が認められた。心エコー検査で非対称性中隔肥大が認められた。この症候群では先天性心異常は非常にまれである。この所見は、心臓の発生に関与する遺伝子座または遺伝子座がこの染色体領域にあることを示唆している。まれではあるが、表現型異常と18p欠失症候群に適合する遺伝学的結果を有する患者を評価する際には、心肥大を念頭に置くべきである。
https://pubmed.ncbi.nlm.nih.gov/35707779/18p欠失症候群患者4人の知的・適応的・行動的特徴
背景
18p欠失症候群のような染色体異常によって引き起こされる症候群において、行動表現型評価と細胞遺伝学的特徴の関連は、遺伝子型-表現型相関の理解を深める可能性がある。
方法
我々は、細胞遺伝学的手法と詳細な神経心理学的評価によって特徴づけられた18p欠失症候群の4人のブラジル人患者について報告する。知的特性、適応特性、行動特性は、それぞれWechsler’s Scale、Vineland-II Scale、Child Behaviour Checklistを用いて評価した。また、ブラジルの社会階層分類基準で定義された主な養育者の教育レベルや世帯収入などの社会経済的指標も収集し、神経認知の変動における環境要因の寄与の可能性を評価した。
結果
4人中2人に知的障害(IQ<70)がみられた。ウェクスラー尺度の結果から、我々のサンプルでは、非言語的行動の観察に基づく社会的状況の解釈が認知的強みを構成する一方、社会的ルールの判断や、単語知識や流暢な言語能力に関連する言語技能は認知的弱みになりうることが示唆された。適応行動に関しては、Vineland-II Scaleでは、コミュニケーション能力や日常生活能力よりも、運動能力や社会性の領域がよく発達していることが示された。児童行動チェックリスト(Child Behaviour Checklist)に基づく内面化行動の問題を呈した患者は1人だけであった。この結果から、社会経済的地位が患者全体の発達に寄与している可能性も示唆された。
結論
我々の結果は、18p欠失症候群患者の中には平均的な知能を示す者もおり、欠失の大きさや家族の社会経済的状況が認知発達に影響を及ぼす可能性があることを示唆している。
染色体18p欠失症候群の遺伝子型と表現型: 症例シリーズ
理由:18p染色体欠失症候群は、18番染色体の短腕の全部または一部が欠失した症候群である。18p染色体欠失症候群の表現型は、欠失の大きさや切断点、欠失に関与する遺伝子の違いにより個人差が大きい。本症候群の多様で非典型的な臨床像を考えると、本症候群の出生前診断は依然として難題である。
患者の問題
我々は染色体切断点が異なる4例の中国症例を報告した。症例1では、軽度の表現型を持つ女性が重度の奇形胎児を出産した。他の3症例は出生前診断であった。これらの表現型はそれぞれ、核透光性(INT)の増加と非侵襲的出生前診断(NIPT)で示された染色体18p上の欠失と重度の水腎症である。
診断
4例は核型分析とアレイベースの比較ゲノムハイブリダイゼーション(array-CGH)により18p染色体欠失症候群と診断された。
介入
核型分析とアレイベースの比較ゲノムハイブリダイゼーションにより異常染色体を解析した。
結果
症例1と症例2では、18p11.32p11.21に11.51Mbと12.39Mbの欠失が認められた。症例3は18p11.3218p11.23に7.1Mbの欠失を認めた。症例4は18p11.3218p11.22に9.9Mbの欠失を認めた。
今回の報告では、同じ染色体切断点を持つ母親と子孫の表現型が有意に異なることを初めて報告した。さらに、胎児における染色体18p欠失症候群の新しい表現型を発見し、出生前診断においてこの症候群の表現型を充実させることができる。最後に、18p欠失症候群の染色体切断点が異なる個体では、表現型が異なることを示した。一方、18p欠失症候群の染色体上のブレイクポイントが同じ個体でも、表現型が著しく異なる可能性がある。
細胞遺伝学的、分子生物学的、表現型的にみた(新生)変異派生染色体18型患者の特徴と文献レビュー
18pの末端欠失と18qの末端重複に加え、18pの隠微な重複を伴うデノボ18番染色体の患者を紹介する。この女児は、微小後背、口蓋裂、両側口蓋垂、高い口蓋、低い耳、短い首、豊かな頬といった軽度の異形性を有していた。また、H型気管食道瘻があり、手術が必要であった。認知能力と運動能力は遅れていた。核型分析では、18番染色体の短腕に追加分節が認められた。染色体マイクロアレイの結果、18p11.32から18p11.23にかけて7.3-MBの末端欠損、18q21.31から18q23にかけて22.2-MBの末端増加、18p11.22から18p11.21にかけて3.9-MBの間質性増加が認められた。我々は、母親が正常18番染色体、der(18)dup(p11.22p11.21)、der(18)dup(p11.22p11.21)inv(18)(p11.22q21.31)の生殖腺モザイクを有するか、あるいは終末のdel/dupと間質性重複の両方が同時に生じたと仮定している。
https://karger.com/cgr/article-abstract/159/2/74/62594/Cytogenetic-Molecular-and-Phenotypic?redirectedFrom=fulltext両親の18番染色体の周囲転位により、表現型的には正常な18分節性片親性ダイソミー児が生まれた。 本研究では、18番染色体の家族性逆位、inv(18)(p11.31q21.33)について報告する。第一子は、18番染色体の組換え型であるdup(18q)/del(18p)に加え、1本の均衡型18番染色体逆転を受け継いでおり、精神発達遅滞はなかったが、軽度の異形性を有していた。2人目の子供は、2本の組換え18番染色体を受け継いでおり、母親からはdup(18p)/del(18q)の派生18番染色体を、父親からはdup(18q)/del(18p)の派生18番染色体を受け継いでいた。この染色体異常は出生前に検出されたが、2つの異数性は互いに補い合うと考えられたため、家族は逆位以外の部分の一親性ダイソミーが子供の発育に悪影響を及ぼす可能性があることを承知で妊娠を継続することにした。一親不分離はSNPアレイによって確認された。生後20ヶ月まで経過観察された子供は健康で正常である。この症例は、2本の反対側の組換え染色体が互いに補い合い、染色体上の2つのセグメント(一方は母方、他方は父方)についてセグメント性片親性ダイソミーに至った初めての報告例と思われる。
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3083628/組換え18番染色体における親染色体周囲逆転の分子機構と文献レビュー
染色体再配列の多くは、ローコピーリピート(LCR)やセグメント重複(SD)を介した非同型相同組換えによって生じる。組換え18番染色体(rec(18))に関する最近の研究は、診断と臨床表現型に焦点が当てられている。われわれは、出生前の組換え18番染色体の2症例を診断し、核型と染色体マイクロアレイ解析を用いて正確なブレークポイント相互間隔を同定した。ブレイクポイント反復要素の分布特性を解析して再配列のメカニズムを推定し、遺伝的傾向を明らかにするために関連文献を検討した。解析した12家族25妊娠のうち、68%がrec(18)例、24%が自然流産例、8%が正常出産例であった。17例のrec(18)症例では、65%が母体由来、35%が父体由来であった。p11.31のショートアームブレークポイントは10例で報告されたが、ロングアームブレークポイントはq21.3(6例)とq12(4例)にあった。18番染色体における近心逆位のブレークポイントはp11.31、q21.3、q12領域に集中している。18p11.31の再配列は非再発性である。ALU、LINE1、MIRはブレイクポイント領域で濃縮され(18番染色体全体で1.85倍から3.42倍濃縮)、SDとLCRは見られなかった。ALUサブファミリーは2対のブレークポイント間で85.94%と83.01%の配列同一性を示した。小さな反復要素は、組換えと結合したDNA修復過程を媒介し、18番染色体上の再配列を促進する可能性がある。母親の逆位保因者は、出生前のrec(18)家系において異常組換えを起こしやすい。組換え染色体は配偶子形成時に優先的分離を示す可能性がある。
https://pubmed.ncbi.nlm.nih.gov/37161033/18p-/18q+症候群と持続性顕微鏡的血尿を呈したペルー人の小児
第18染色体転座の保因者は、組み換え染色体を持つ子孫を持つ可能性があり、その結果、臨床的に様々な症状を示す患者が生まれる。18p-/18q+再配列の患者は、いくつかの臨床的特徴を共有しているが、他の特徴は異なっている。このような分岐の要因としては、特に逆位セグメントの長さが挙げられる。ここでは、異形、知的障害、持続性顕微鏡的血尿、大動脈仮性動脈瘤、下行大動脈炎などを有するペルー人の小児について述べる。家族の核型分析により、母親が18[inv(18)(p11.2q21.3)]の近心逆位保因者であることが判明した。この子供は組換え18番染色体を保有しており、染色体マイクロアレイ分析により、患者の18番染色体には2つのゲノム不均衡が検出された。18p-/18q+の表現型では、持続性顕微鏡的血尿は報告されていない。本症例は、このタイプの組換え18番染色体におけるヘミ接合性に関連する遺伝子型-表現型相関を満たさない、あるいは患者が高齢になるにつれてこのような特徴を呈するという、まだ知られていない重要な役割を他の因子が担っていることを解明している。
https://pubmed.ncbi.nlm.nih.gov/29142771/微小欠失・重複
微小欠失Microdeletions: 5025人に1人・重複Duplications: 14286人に1人
最近の研究では、報告されたすべての染色体異常の4.7%が、微小欠失を含む欠失であることが示され、出生10,000人あたり1.99人の有病率が得られた(3)。 重複はさらに少なく、出生10,000人あたり0.7人の有病率を示し、報告された全染色体異常の1.6%に相当する(3)。
https://www.sciencedirect.com/science/article/pii/S0015028216629643常染色体劣性疾患
常染色体劣性の障害
常染色体劣性(AR)疾患は、かなりの遺伝性疾患の一部を構成しており、相当な疾病負担の原因である。新生児1000例で約1.75-5人に影響する(これに対し、常染色体優性疾患では1000例で1.4人)。
https://www.nature.com/articles/s41525-021-00203-x全染色体の異数性および部分欠失部分重複の検査をする必要はないのか?
認証施設は21,18,13トリソミーのみの検査を推奨しています。ほかの検査は不要であるということをよく耳にします。ヒロクリニックはそれにもかかわらず、その他の様々な検査を提供しています。なぜなら、これらの検査結果は頻度が総合すると頻度が低いのものではなく、母体に影響を当たるからです。その頻度、影響については認証施設では検査そのものをやっていないので、実際のデータがありません。(公表していないだけかもしれません。)21,18,13番以外の異数性、部分欠失部分重複のデータはヒロクリニックでは統計を取っているので存在しますので、これらを他国のものと比較してみたいと思います。以下がヒロクリニックでの抜粋した検査結果です。
海外の情報は上記のサイトから取得してみたいと思います。これは専門家のみが見れるサイトなので、簡単に訳してみたいと思います。(誰でも見れますが、専門用語が多いので難しいです)
①まずはトップページです。翻訳したものを載せます。我々の使っている表現が異なるところは赤字で補足させていただきました。その後に各国で報告されているダイジェストを載せます。いずれもヒロクリニックのデート大きな差がありません。
公的資金による全国一次非侵襲的出生前スクリーニングの結果
ベルギーは、すべての妊婦を対象とした一次スクリーニングとして NIPT を実施し、全額償還した最初の国です。この出版物では、ベルギーのすべての遺伝センターで構成されるコンソーシアムが、すべての妊婦に一次検査オプションとして拡張 NIPT を提供した 2 年間の臨床経験について報告しています。スクリーニングされた153,575人の妊娠のうち、まれな常染色体トリソミーと部分欠失/重複がそれぞれ0.23%と0.07%の症例で見つかりました。侵襲的な診断的産科処置は 52% 減少しました。著者らは、拡張NIPTアプローチがベルギーの出生前ケアにうまく導入されており、NIPTを提供する他の国々の枠組みとして機能する可能性があると結論付けている。
③オランダです。
ゲノムワイドNIPSで検出された共通トリソミー以外の染色体異常の起源と臨床的関連性:TRIDENT研究の結果
この出版物は、拡張 NIPT を使用して一般的な異数性以外の染色体異常を検出する潜在的な臨床的有用性を実証しました。この研究集団では、拡張 NIPT を受けた症例の 1.6% が、まれな常染色体トリソミーまたは大きな部分的欠失/重複である稀な染色体異常のスクリーニング陽性でした。これらの症例のうち、60% は、異常な胎児表現型や子宮内発育制限などの有害な妊娠転帰と関連していました。
④アメリカです。
ゲノムワイドな無細胞 DNA スクリーニング: コピー数変異に焦点を当てる
この出版物は、86,902 件の拡張 NIPT サンプルのコホートのうち、少なくとも 1 つの染色体部分欠失/重複のスクリーニング陽性となった 490 件 (0.56%) の症例に焦点を当てています。これらの症例の 50% で診断結果が得られ、PPV が 70% を超えることが判明しました。
⑤中国です。
拡大染色体疾患症候群に対する非侵襲的出生前スクリーニングの臨床的有用性
この出版物では、オールリスク妊娠集団における異数性およびゲノム全体の微小欠失/微小重複症候群の両方に対する拡張 NIPT の臨床成績を検証しました。単胎妊娠の患者94,085人のコホートが前向きに登録された。この拡張 NIPT では、サンプルの 1.2% で臨床的に重大な胎児染色体異常が検出され、スクリーニング陽性異常に対するそれぞれの PPV が計算されました。


Copyright (c) NIPT Hiro Clinic All Rights Reserved.