

アレッサンドラ・ディ・ノーラ1、 ジェルマーナ・レナ1、 アンドレア・ジューン1、 アレッシア・ディ・マリ2、 ピエルイジ・スミラリ3、 カルメロ・ミナーディ4、 ピエロ・パヴォーネ3
1カタニア大学、イタリア、カタニア大学、小児科、院後研修プログラム、臨床・実験医学部
2カタニア大学、イタリア、カタニア大学放射線学部、大学院後研修プログラム、放射線科
3イタリアカタニア大学小児神経内科
4イタリア、カタニアの大学病院「G. Rodolico」麻酔・集中治療部
グローバル・メッド・ジェネット
通信宛先
Alessandra Di Nora医師、病院ポリクリニック、SanoMarco、カタニア大学、Via S. Sofia 78、95123 Catania、イタリア
(電子メール: alessandradinora@gmail.com)
要約
発達遅滞および神経学的障害のある小児では、診断は幅広い原因のため困難である。 過去10年間から、アレイ比較ゲノムハイブリダイゼーション(CGH)の使用は、複雑な表現型の診断を得るために大きな貢献を提供した。 7番染色体は、染色体異常、再配列、および臨床症状を含む欠失と頻繁に関連するため、医学遺伝学に関心が集まっている。本稿では、重度の神経発達遅滞、頭蓋顔面異型、および肺狭窄を有する3歳男児を報告する。このアレイCGH解析は、7番染色体上の14.4Mb(7q21.3~7q31.1)の重複を明らかにした。 現在までの文献をレビューすることにより、著者らは、以前には報告されていなかったこの再配列に関連する神経学的および異形奇形について初めて報告した。
キーワード
7q21.3-q31.1、直列CGH、発生遅延、顔の特徴、重複
はじめに
長腕(q)が重複している個体は稀にしか報告されていない。孤立した “純粋な “7q型は、長腕全体、あるいは染色体セグメントの間質部、近位部、遠位部のいずれかに関与する染色体異常によって分類されています。1,2,18 Novalesら14は、7q染色体のセグメントに応じて罹患者を区別し、次のような結果を得ました。7q22から7q31の間質性重複を有する者は、前頭隆起、長い睫毛、狭い口蓋裂、眉毛、多毛、小さい鼻、長い上唇、眼球障害からなる顔の特徴を示し、骨格異常、口蓋裂、早死にを伴わない。7q31から7qterの重複者は、発達遅延を示し、顔の特徴としては、大きなフォントアネル、狭い口蓋裂、多毛、小さい鼻、口蓋裂、小顎症、低位置で奇形の耳などがあり、さらに、骨格の異常と早期死亡も記録された。7q32から7terの重複は、発達遅延、小さい鼻、低い位置にある耳を特徴とし、小顎症や口蓋裂はなく、骨格の異常、神経症状、早期死亡が見られた。
本論文では,7番染色体の長腕が7q21.3からq31.1まで14.4Mbにわたって純粋に部分重複している3歳の男児を報告した。本症例で認められた臨床症状を報告し,遠位のセグメントが関与している子供の臨床的特徴と比較し,さらに間質性の7番染色体セグメントが関与している子供の臨床的特徴と比較して,罹患した各セグメントに特異的な臨床症状の可能性を明らかにすることを目的としている。
症例提示
この3歳の男性患者は、健康で、血縁関係のないイタリア人の両親の3番目の子供である。 2人の兄弟姉妹(5歳姉妹、7歳兄弟)は健康で、遺伝性疾患の家族歴はない。 妊娠時,母は32歳,父は35歳であった。 母親は妊娠中に感染症にかかったことを否定し、タバコを吸ったり、妊娠中に薬を飲んだことがない。 子宮内超音波検査では7か月までに発育性狭窄を示し、母親は胎動が減少したことを認めた。 子供は妊娠39週に骨盤位のため帝王切開により出産した。 出生時体重は2,350g(3番目のパーセンタイル)、長さ48cm(10番目のパーセンタイル)、後頭前頭周囲(OFC)33cm(3番目のパーセンタイル)であった。 生後2日目に、小児は呼吸窮迫に苦しみ、治療および検査のためにイタリア、カタニア大学小児クリニックの新生児集中治療室に入院した。
頭部超音波は脳室周囲白質の軽度の低密度を示した。 心エコーは心房中隔欠損を伴う肺動脈弁狭窄を示した。 生後10日目、患者は速やかな心臓インターベンションの適応なしに良好な状態で退院した。 最初の1か月の間、患者の臨床像は、主に軽微な顔面の特徴と、特に筋緊張低下に関する発達のマイルストーンの遅れによって特徴づけられた。 2.5歳時、小児は発達遅滞および頭蓋顔面奇形による検診のためイタリアのカタニア大学小児科に入院した。 この年齢では、体重は12kg(10パーセンタイル)、長さ88cm(10パーセンタイル)、OFC 47cm(3パーセンタイル)であった。 「形態学的要素:標準用語:Am J Med Genet 2009, 149A (1): 1-127」によれば、小頭症、タール脳症、顕著な前頭隆起、内側部で剃毛した弓状の眉、内側上角のひだ、下向きの眼瞼裂、平坦な鼻橋、丸い先端と前傾した鼻孔を有する鼻の薄い、平坦なフィルター、薄い口唇、回転軸を有する低位の埋め込み耳、および細い毛髪からなる頭蓋顔面の特徴を示した。
筋肉腫は低緊張性で、皮下組織は不良であった。 患者は、足首弁狭窄、関節拘縮における膝、キール胸、肩甲翼、手関節弛緩、および腹直筋ジアステーシスを示した。発達遅滞が出席し、非常に重い言葉を発することができた。 歩行は不安定だった。 膝蓋骨骨骨性骨壊死反射消失は不適格。 収縮期マウス2/6 Levineは顕著であり、心エコーは心房中隔欠損を伴う肺動脈弁狭窄の存在を確認した。 ルーチンの検査室分析電解質、血漿および尿中アミノ酸、甲状腺マーカー、有機酸、血漿プリン、トランスフェリン等電点電気泳動で左停留精巣が認められ、総コレステロールは正常範囲内であった。 覚醒時および睡眠時の脳波は正常であった。 拡散強調画像シーケンスを用いた脳磁気共鳴画像法(MRI)は、テント下およびテント上構造および脳室の規則的な容積および解剖学を示した。 脊椎MRIでは、S5後弓に裂け目、S3からS5にかけて仙骨遠位部脂肪腫症、右S2に小さな根尖周囲嚢胞Tarlov型が指摘された(図1、2参照)。
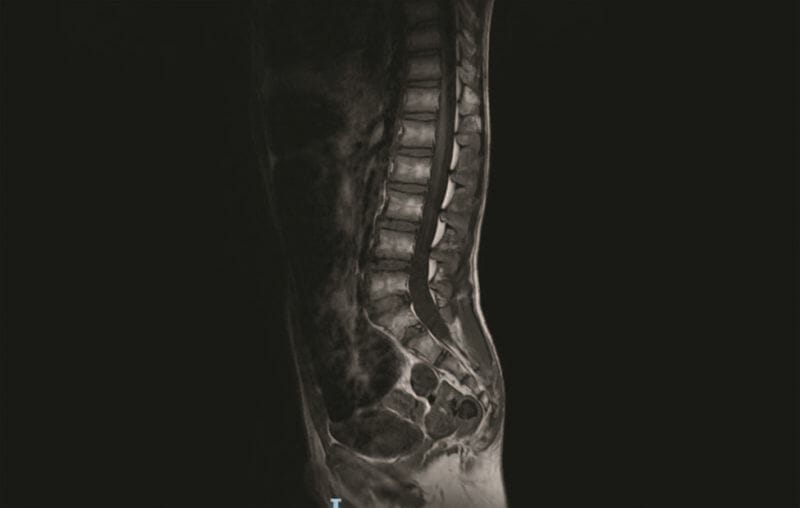
図2 S2心尖部周囲嚢胞を示すスピン磁気共鳴画像法。
遺伝子検査
ヒト細胞遺伝学的命名法のための国際システム11 によれば、小児およびその両親のリンパ球培養後に高精度核型分析を行ったところ、平均解像度は550バンドであり、分子細胞遺伝学には細胞塗布プローブを用いた。
Cytofure ISCA 8×60K v.2(蛋白質O‐GicNacTransferase,OGT)を用いたアレイ比較ゲノムハイブリダイゼーション(CGH)(ヒトDNA,Promega)分析を小児とその両親からの末梢血試料から抽出したメドンゲネティックDNAを行った。 ソフトウェア解析は、Cytofure Analysis Software(カリフォルニア大学サンタクルーズ校ゲノムアセンブリ19)で行った。 品質スコア(デリバティブ・ログ・レシオ・スプレッド、DLRS)は<0.25であった。 分析パラメータ:4つの連続するプローブ。 分解能:50~100Kb。 RASopathiesの次世代シークエンシング(NGS)パネルを実施した。
結果
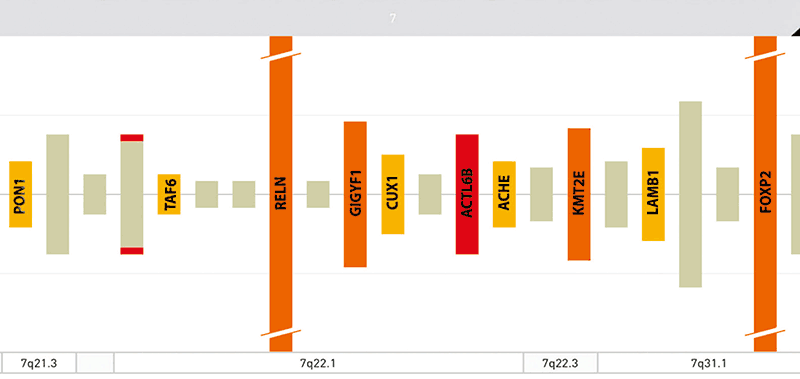
分析は、子供において、染色体7の長腕の染色体内部分重複を伴う46,XY核型を示し、14.4Mb(7q21.3-q31.1):arr[hg 18](以下、図3A,3B)に及んだ。 重複のセントロメアマージンは、95054968pb位の正常オリゴマーおよび109476740pb位の重複オリゴマーによってなされる。 109557905pbの正常オリゴマーと109476740bpの重複オリゴマーによって作製されたテロメリックマージン(図4)。 RASopathies NGS解析では、変化は認められなかった。 両親の核型およびアレイCGHは正常であった。
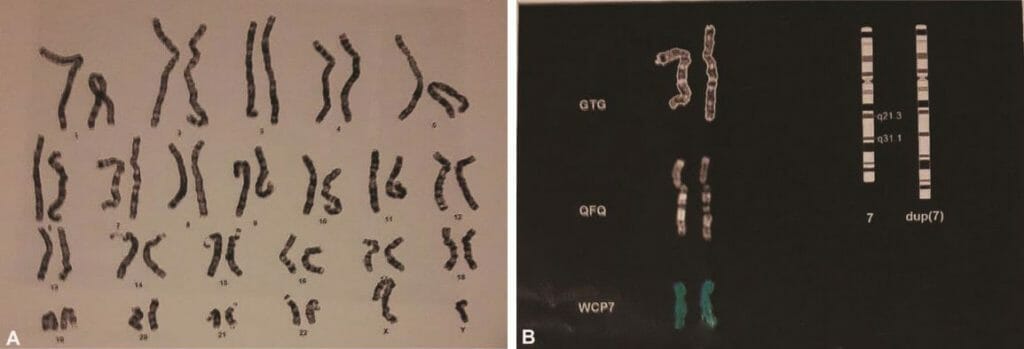
図4 再構成領域に含まれる遺伝子を示すOMIMから改変されたイメージング
考察
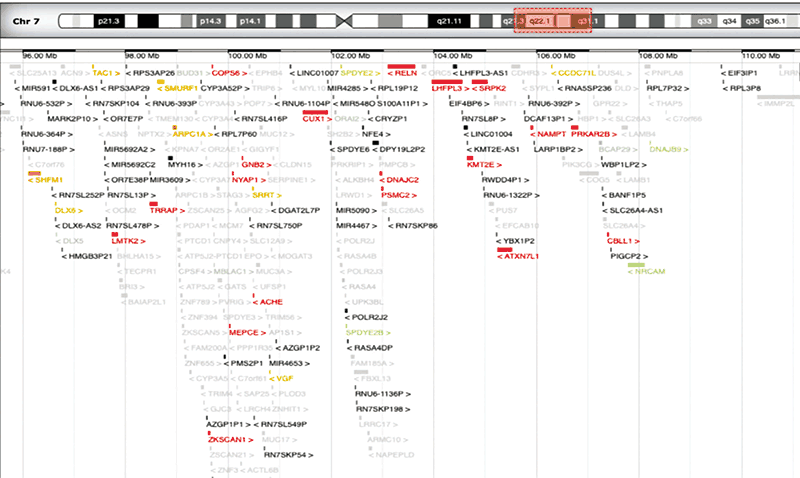
小児は、軽度の非特異的頭蓋顔面特徴、小頭症、軽度発達遅滞、心臓および骨格障害からなる先天異常の複合体を示した。 生殖器、手、腹部も侵された。 先天性心疾患は中隔欠損を伴う肺動脈弁狭窄からなり、骨格異常はS5後弓裂、S3~S5の遠位仙骨脂肪腫症、および右S2心尖周囲嚢胞Tarlov型を示した;関節拘縮および足首弁狭窄における膝も存在した。 さらに,腹直筋ジアスタシス,左停留精巣,手関節弛緩を認めた。 アレイCGHは、いくつかのOMIMおよびRefSeq遺伝子を有する染色体7q21.q31.1上の14.4Mbの大きなゲノム重複を示した:再編成された領域は230以上の遺伝子を含む(図4)。 SFARI遺伝子データベースによれば、ACTL6B、FOXP2、PONI、KMT2Eなどのいくつかのものは、シナプス可塑性および自閉性シグナル伝達機構ならびに脳および他の臓器に影響を及ぼす先天性疾患の原因となる他の構造的異常(図5)に関与している。
図5 自閉症スペクトル障害に関与する遺伝子を示すSFARI遺伝子を改変した画像。
染色体7qは異なる構造再編成を受けやすい。 浸潤した間質、近位、または遠位の各セグメントに応じた特異的な臨床徴候を強調しようとする試みは、明確に定義されていない。 Alfonsiらは、q21.1-q22.3領域が関与する部分的な染色体7q重複を呈する観察例の臨床的特徴を、類似した7q再構成を有する文献から抽出した6人の患者と比較することを試みた。 これらの著者らは、知能遅延と低集合奇形耳の存在が、それら自身を含むすべての症例で認められ、眼の斜視と前頭突起の特徴が6例、骨格異常が5例で認められた。 患者のうち3人は、生殖器欠損、巨頭症、及び高眼瞼症、小さな上向き鼻、小顎後症、口蓋裂、及び高弓状口蓋のようないくつかの類似した頭蓋顔面の特徴を示した。1 著者らは、純粋な染色体7q近位断片を有する患者では、特異的な診断上の臨床的特徴が生じていないという結論を導いた。1 本研究では、本症例を含む文献で報告された7q重複間質性(7q22~7q32)セグメントの8症例の各々の臨床データを分析し、臨床的類似性または差異を評価した(表1)。 間質性群では、男性3例、女性4例が罹患しており、性別の有病率は認められなかった。発達遅滞および軽度の非特異的顔貌が全症例で報告された。 特に、いくつかの顔の特徴を見ると、前頭隆起とエピカナスは7例中6例、低位耳は4例、小顎後症は2例で報告された。 低血圧、発育遅延は7例中6例で認められ、眼内斜視は5例であった。 3人の小児は小頭症であり、心臓、骨格、生殖器の異常発汗が本症例のみで報告されたが、他の小児では報告されなかった。 ヒルシューティズムと股関節亜脱臼は1例で報告され,脳奇形と腎異形成は1例のみであった。2, 4, 7, 15, 17, 18, 21 これらの結果を分析すると、知能遅延および軽度の頭蓋顔面遅延を除き、明確な診断を示す特異的な臨床徴候は提起されていないことが明らかである。
純粋な間質性7q重複と純粋な間質性7q重複の臨床データを、文献で報告された6例と現在の小児で観察された6例とを比較して、検討した(表2)。3, 6, 10, 17, 20, 22 これらの結果を分析すると、わずかな非特異的頭蓋顔面の特徴、知的遅延、およびわずかな頻度での骨格障害が、遠位および間質性染色体7q浸潤を伴う現在の小児群のいずれにおいても、最も代表的な臨床徴候であることが明らかであるように思われる。
表2 報告された6例(8例、11例-15例)と本症例の純粋な間質性7q重複の臨床データ
| グレース他(1972) | バーガー他(1974) | セルビル他(1975) | ローマ他(1990) | メガルバネ他(2000) | ワイマー他(2011) | 現在の症例 | 合計 | |
|---|---|---|---|---|---|---|---|---|
| 染色体7 | q22-q32 | q21-q31 | q22-q31 | q22-q31.2 | q22-q31.3 | q21.1-q31.3 | q21.3-q31.1 | |
| 性 | F | M | M | F | F | F | M | 3 M/4 F |
| 顔の特徴 | ||||||||
| 前面ボス加工 | – | + | + | + | + | + | + | 6/7 |
| エピカントゥス | + | + | + | + | + | + | + | 6/7 |
| 小顎後症 | – | – | – | – | + | – | + | 2/7 |
| 低音 | + | – | + | – | + | – | + | 4/7 |
| 開発遅延 | + | + | + | + | + | + | + | 7/7 |
| 小脳症 | – | – | – | – | + | + | + | 3/7 |
| 低血圧 | – | + | + | + | + | + | + | 6/7 |
| 発育遅延 | + | – | + | + | + | + | + | 6/7 |
| 先天性心疾患 | – | – | – | – | – | – | + | 1/7 |
| 性器奇形 | – | – | – | – | – | – | + | 1/7 |
| 骨格異常 | – | – | – | – | – | – | + | 1/7 |
| 眼の異常 | – | + | + | + | + | + | – | 5/7 |
| その他の異常 | – | + | + | + | + | + | + | 5/7 |
注:その他の異常としては、2例(14、15)で報告されている多毛症、1例8で報告されている難聴と腎異形成、そして本症例で報告されている脊椎の異常があります。
純粋な単離された7q染色体の重複を除いて、7q異常は、同じ染色体または他の染色体が関与する再配列と関連して起こり、異なるより複雑な臨床症状を引き起こすことがある。 Fruhmesserらは、7q31.2.2q32.3の追加的反転を伴う7q22q32の部分的トリソミーの症例を報告した。15 小児は発達遅滞,発育不全,手の異常,動脈管開存と卵円孔開存を呈する先天性心疾患を示した。
頭蓋顔面の特徴は、前頭部隆起、わずかに下向きの眼瞼裂、広い鼻根、長い親和性の小顎症、および突出した耳からなっていた。 Pathtinigeらは、7q22.-qterの女児トリソミーで、14番染色体上の遺伝物質の喪失が最小限であることを報告した。13 子宮内発育遅延、発達遅滞、および脳室肥大は、非対称性頭蓋、三角形顔形、高額、高頭蓋症、前頭葉症、平坦な鼻橋、小顎症、および低位奇形耳を呈する頭蓋顔面の特徴を伴う主な徴候であった。16 著者らは、この症例では遺伝子型と表現型の明確な相関は認められなかったという文献の結果と一致した。
本患児は肺動脈弁狭窄から成る先天性心疾患を示し、これはヌーナン症候群の典型的な徴候の1つとして報告されている異常である。 この症候群は、RAS/MAPK経路17 の共通の調節異常を共有する「RASopathies」と呼ばれる疾患群に分類されるため、RAS/MAPK経路に関与する変化と関連している。 心臓欠損を含む現在の小児によくみられる臨床徴候のため、我々はRASopathy主要遺伝子のためのNGSパネルを用いた遺伝学的研究を拡張し、この症例では陰性結果が得られた。 さらに、本患児は骨格系、特にS5後弓裂を伴う脊椎、仙尾骨脂肪腫症、および仙尾骨奇形に影響する異常を示した。 異常(最後のもの)は、Currarino症候群(CS)の一部である。
常染色体顕性仙骨無形成として知られるCSは、仙骨の部分的欠損を特徴とする古典的な三徴であり、第一仙椎の損傷、仙骨前部腫瘤、直腸肛門異常を伴う。18 CSは、尾部のみの奇形から複雑な臓器系奇形まで様々な臨床的発現を示すことがある。 この疾患は、染色体7q368にマップされたホメオボックス遺伝子であるHLXB9の有害な変異体に関連している。 Pavoneらは、小頭症、皮質肥厚の欠如、下部虫部低形成、CSの徴候を伴う出生前および出生後の発育遅延および複雑な奇形を呈する女児が、仙骨部分無形成および完全尾骨無形成、仙骨内類皮腫、硬膜内脂肪腫、異所性肛門、および繋留索として認められたことを報告している。19-22 小児は7q34-q35のde novo重複および7q36の8.8-Mb欠失を示した。 この配列にはHLXB9(CS)遺伝子が含まれ,Pavone et alalの症例報告は見つからなかった。14 今回の小児で報告された脊椎奇形とCSで報告された脊椎奇形との間の臨床的関係は未だ確立されていない。
結論
結論として、q7重複の異なる近位部、間質部、遠位部の純セグメントに関与する個人に認められる臨床徴候および身体器官は、非特異的であり、診断的には示唆されない。 特有の顔像および軽度の発達遅滞非特異的な診断徴候が報告されていないことを除いて、浸潤した間質分節に浸潤した1例についても、同じ結果が得られた。
倫理的承認
本試験はヘルシンキ宣言の倫理指針に準拠し、イタリア、カタニアの大学病院倫理委員会「ポリクリノ・ガスペア・ロドリコ」(nd: 1268 23/04/2017)により承認された。 本症例報告書および付随する画像を発表するために、患者から文書によるインフォームド・コンセントを得た。
著者の貢献
各著者は、著作物の構想またはデザインに実質的な貢献をし、重要な知的内容のためにそれを批判的に改訂した。 さらに、各著者は最終版を公表することを承認した。 逆に、各執筆者は、作業のいかなる部分の正確性又は完全性に関する問題も適切に調査され、解決されることを確保する上で、作業のすべての側面について説明責任を負うことに合意した。
資金調達
なし。
利益相反
申告なし。
謝辞
調査にご参加いただき、資料・画像の公表にご同意いただき、ありがとうございました。
参考文献
1 Alfonsi M, Palka C, Morizio E, et al. 純粋な部分7q重複の新しい症例。 細胞遺伝学ゲノム研究2012;136(01):1-5
2 Bartsch O, Kalbe U, Ngo TK, Lettau R, Schwinger E. 部分重複7qの臨床診断。 Am J Med Genet 1990;37 (02):254–257
3バーガー・R、デラー・J、オルティスマサチューセッツ州。 第7染色体の長さでは、トリソミーが少なくなる。 Nouv Presse Med 1974;3(29):1801–1804
4 CouzinDA,HaitesN,Watt JL, JohnstonAW. 2人の小児における7(q32–qter)部分トリソミー症候群。 JMed Genet 1986;23(05):461-465
5 Fruhmesser A, ErdelM, DubaHC, et al.CombinedDup(7)(q22.1q32.2), Inv(7)(q31.31q31.33), およびIns(7;19)(q22.1;p13.2p13.2)は、12歳の男児で、発達遅延および種々の異形を伴う。 Eur J Med Genet 2013;56(07):383–388
6グレースE, Sutherland GR, Bain AD. 家族性挿入転座。 Lancet 1972;2(7770):231
7 Keith CG, Webb GC, Rogers JG. 染色体セグメント7q32–q34の重複と関連する外側直筋の欠損。 J Med Genet 1988;25(02):122-125
8 Kim IS, Oh SY, Choi SJ, et al. カラリノ症候群の韓国人患者におけるHLXB9遺伝子の臨床的および遺伝的解析 J Hum Genet 2007;52(08):698-701
9 Lynch SA, Wang Y, Strachan T, Burn J, Lindsay S. 常染色体顕性仙骨無発生: Currarino症候群。 J Med Genet 2000;37(08):561-566
10 Mégarbané A, Gosset P, Souraty N, et al. 染色体7q22-q31の重複:新規症例の報告とレビュー。 Am J Med Genet 2000;95(02):164–168
11 Mcgowan-Jordan J, Simons A, Schmid M, eds. ISCN 2016:ヒト細胞ゲノム命名法のための国際システム。 バーゼル:S. Karger; 2016
12 NovalesMA, Fernandez-Novoa C, Hevia A, San Martin V, Galera H. Partial trisomy for the long armof chromosome 7. 症例報告およびレビュー。 Hum Genet 1982;62(04):378-381
13 Paththinige CS, Sirisena ND, Kariyawasam UGIU, et al. 部分的トリソミー7q22.1による多発性先天異常の小児! 母性遺伝性バランス転座の結果として生じるqter:症例報告および文献レビュー。 BMC Med Genomics 2018;11(01):44
14 Pavone P, Ruggieri M, Lombardo I, et al. 小脳症、感音難聴、および遠位7qの重複欠失を伴うCurrarino三徴。 Eur J Pediatr 2010;169(04):475–481
15 Pavone P, Corsello G, Marino SD, RuggieriM, Falsaperla R. 7q31.32部分重複: 変形、自閉症スペクトラム障害、中等度の知的障害、およびてんかんの小児の最初の報告。 文献レビュー。 てんかん研究2019;158:106223
16 Roberts AE, Allanson JE, Tartaglia M, Gelb BD. ヌーナン症候群。 Lancet 2013;381(9863):333-342
17 Romain DR, Cairney H, Stewart D, et al. 7番染色体のまれな構造的再編成による7q部分トリソミーの3例。 J Med Genet 1990;27(02):109-113
18 Scelsa B, Bedeschi FM, Guerneri S, Larta F, Introvini P. Partial trisomy of 7q: 症例報告と文献レビュー。 J Child Neurol 2008;23(05):572–579
19 Schubbert S, Shannon K, Bollag G. 発達障害および癌における高活性Ras.Nat Rev Cancer 2007;7(04):295-308
20 Serville F, Broustet A, Sandler B, Bourdeau MJ, LeloupM. トリソミー7qパーティセル Ann Genet 1975;18(01):67-70
21 Verma RS, Conte RA, Pitter JH. 第7染色体長腕の末端バンドの縦列重複(二重複(7)(q36-qter))。 J Med Genet 1992;29(05):344-345
22 Weimer J, Heidemann S, von Kaisenberg CS, et al. 7番染色体、9番染色体、10番染色体が関与する複雑な家族性再配列の結果、7q21.2-31.31のトリソミーが単離された。 Mol Cytogenet 2011;4:28
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。


