
現在は国内検査を行なっておりません。
海外に発送しておりますので、ご相談ください。
70%の両親がキャリア?潜性遺伝子検査の有用性について
70%の人が何らかの遺伝子の異常(変異)を持っています。
このような遺伝子の異常を持つ人は「キャリア(保因者)」と呼ばれます。
見た目や体に異常がなくても、その遺伝子が子どもに影響を及ぼす可能性があります。
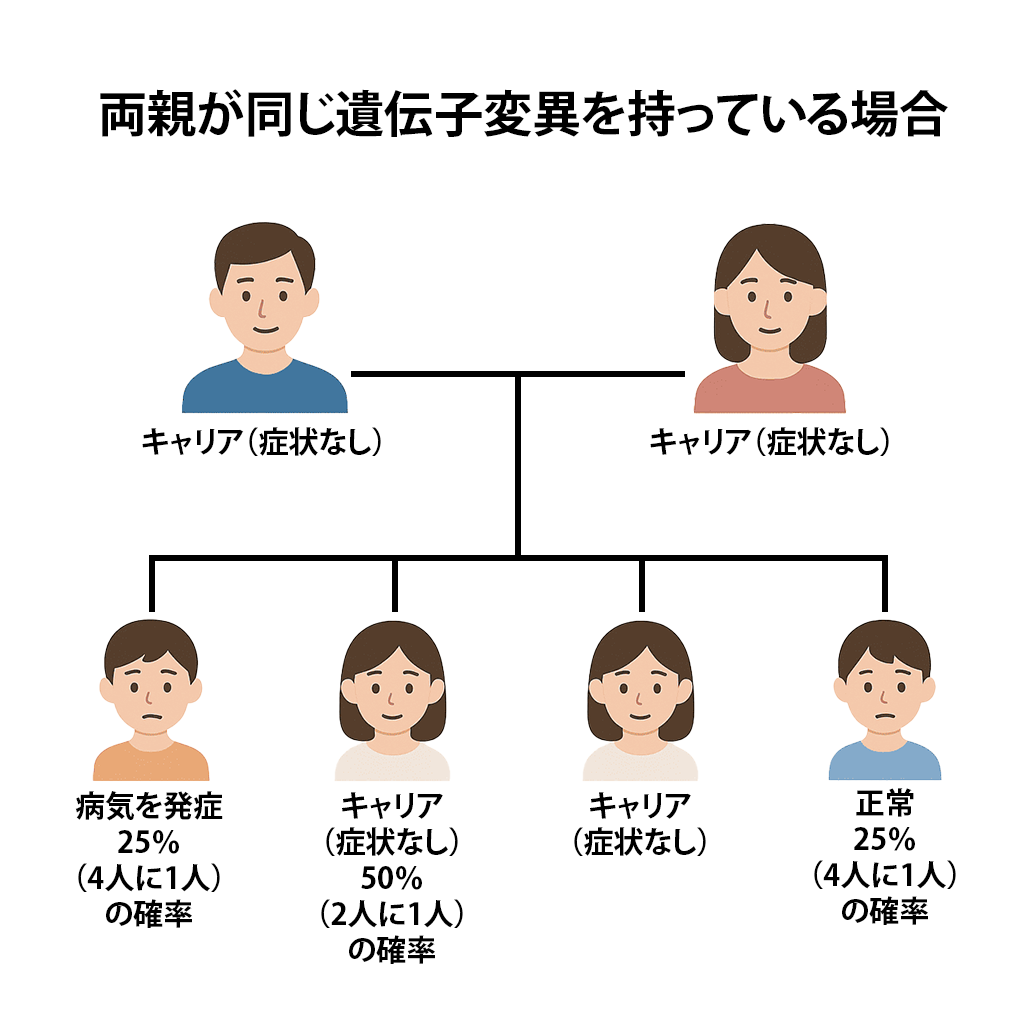
両親が同じ遺伝子の変異を持っている場合、その子どもが病気を発症する確率は25%、キャリアとなる確率は50%、正常な遺伝子を受け継ぐ確率は25%です。


では、そのような人にどのような検査が行われるのでしょうか。また、どのような疾患が見つかるのでしょうか。

①ヒロクリニックのキャリアスクリーニングテスト231とは?
ヒロクリニックでは、出生前に胎児に関連する最大231種類の重篤な潜性遺伝疾患に関連する遺伝子を調べることができる検査を提供しています。この検査は、米国産科婦人科学会(ACOG)や米国人類遺伝学会(ASHG)でも広く情報提供すべきとされている、国際的にも重要視されている検査です。
潜性遺伝疾患とは、父親と母親の両方が同じ種類の遺伝子の変異を持っているときに、子どもに発症する可能性のある病気です。この検査では、父親と母親の口腔粘膜(頬の内側)から遺伝子を採取して調べ、両親が同じ変異を持っていないかを確認します。

ヒロクリニックの調査では、約70%の人が1つ以上の潜性遺伝子変異を保有していることがわかっています。これは「キャリア(保因者)」と呼ばれ、見た目や体の調子に問題はなくても、子どもに遺伝子を伝える可能性がある状態です。
両親が同じ遺伝子変異を持っている場合、次のような確率で子どもが病気を発症するリスクがあります。
- 25%(4人に1人)の確率で病気を発症
- 50%(2人に1人)の確率でキャリア(症状なし)
- 25%(4人に1人)の確率で正常
この検査でリスクが高いとわかった場合は、確定検査として羊水検査が推奨されることがあります。
②遺伝子の“変異”が病気を引き起こすことがある
私たちの体は、「遺伝子」という設計図のような情報にしたがって作られています。遺伝子はおよそ2万個あり、親から子へと受け継がれます。髪の色や背の高さ、体質など、さまざまな特徴を決める大切な情報です。
ところが、まれにこの設計図に変異が生じることがあります。この変異が原因で、特定の病気になることがあるのです。
たとえば「潜性遺伝」という仕組みで起きる病気は、父親と母親の両方から同じタイプの遺伝子の変異をもらったときにだけ発症します。片方の親だけが変異を持っていても、本人は潜性遺伝疾患の病気にはなりません。
潜性遺伝の病気の一例として、15番染色体にある「OCA2」という遺伝子の変異があります。この変異を父親と母親の両方から1個ずつ、合わせて2個受け継ぐと、「Pタンパク質」という、肌や髪、目の色を決める「メラニン」という色素を作るのに必要なタンパク質が作られなくなります。その結果、メラニンが作られず、肌や髪の色が白くなる「眼皮膚白皮症(Oculocutaneous Albinism)」という病気になります。

また、「X染色体」という性別に関係する遺伝子の異常による病気(X連鎖潜性疾患)は、男の子に発症しやすいのが特徴です。男性はX染色体を1本しか持っていないため、そこに変異があると代わりがなく、病気が出やすくなります。一方、女性はX染色体を2本持っており、どちらか一方に異常があっても、もう一方が正常であればその働きを補うことができるため、発症しにくい傾向があります。
③誰にでも起こりうる遺伝性疾患
潜性遺伝の病気は珍しいと思われがちですが、実は3000種類以上もあります。すべての病気をあわせると、100組の夫婦のうち1〜2組に、遺伝性疾患を持つ子どもが生まれる可能性があるとされています。つまり、誰にでも関係のある話なのです。
そのため、妊娠前に検査を受けておくことで、万が一リスクが見つかった場合にも冷静に対策を講じることができます。たとえば、体外受精で健康な胚を選ぶ、妊娠後に羊水検査をして赤ちゃんの状態を確認する、などの選択肢があります。
④母親と子どもの健康管理にも役立つ
キャリアスクリーニングテスト231は子どもの病気のリスクを知るだけでなく、母親の健康管理にも役立ちます。たとえば、妊娠中に注意が必要な出血のしやすさや、心臓の病気のリスクなどを前もって知ることができれば、医師も事前に準備ができます。
このようにキャリアスクリーニングテスト231は、安心して妊娠・出産するためのサポートにもなります。ヒロクリニックでは、NIPT(非侵襲的出生前診断)との併用も可能で、NIPTを受検された方には、キャリアスクリーニングテスト231をオプション価格で提供しています。もちろん、NIPTを受けていない方も単独で受検できます。
⑤日本人に特有なリスクもある
日本では、いとこ同士の結婚が多かった時代があったため、それによって特定の遺伝子の変異が集団の中に残りやすくなっています。そのため、日本人に多い病気もあります。
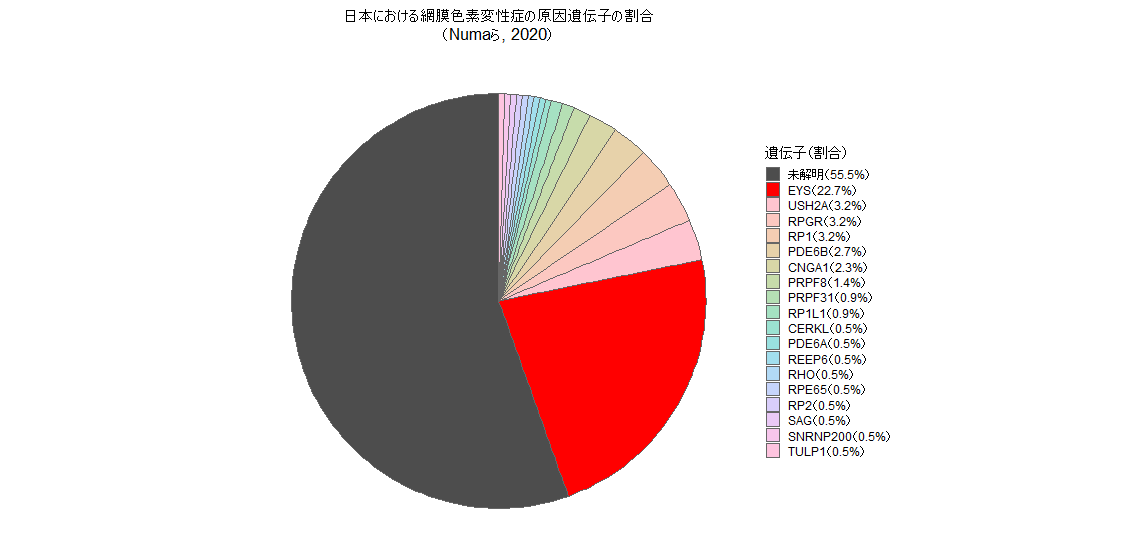
たとえば、「網膜色素変性症」や「白点眼底症」など、目に関わる遺伝病の中には、日本人に特に多いものがあることがわかってきました。特に、網膜色素変性症の原因遺伝子としては「EYS」が日本人において最も高頻度で関与していることが明らかになっています。これらの病気は特定の遺伝子変異によって起こることが多く、意外と身近な疾患です。
このような日本人に特有の遺伝子の変異は、海外のデータには載っていないこともあります。そのため、日本人向けに作られたキャリアスクリーニングテスト231が非常に重要です。
⑥未来の医療へ──公平に、誰もが選べる社会に
これからの医療は、「病気になってから治す」のではなく、「病気を防ぐ」ことが大切になっていきます。キャリアスクリーニングテスト231はそのための第一歩です。
オーストラリアやオランダでは、潜性遺伝子検査を国の医療制度に取り入れる取り組みが進んでいます。日本でも、すべてのカップルが公平に検査を受けられるよう、保険適用が可能な制度づくりが求められています。
ヒロクリニックでは、妊娠前のブライダルチェックとしても、カップルでのキャリアスクリーニングテスト231を推奨しています。これにより、これからの暮らしの選択において重要な判断材料を早めに得ることができ、安心して出産に臨む準備ができます。
さらに、出生後の赤ちゃんにもキャリアスクリーニングテスト231を行うことで、将来の健康リスクを早期に発見し、個別に対応した健康管理が可能になります。頬の内側をこすって検体を採取するだけの、痛みのない方法で実施できます。
この検査は将来生まれてくる命を守るための「知る」という選択肢です。米国の主要な学会でも推奨されているこの検査を、日本でも多くの人に正しく伝え、選べる社会を目指していくことが、これからの医療に求められています。
参考・引用文献
- Li, Huanyun, et al. ‘P806: Application Value of Noninvasive Prenatal Diagnosis of Recessive Monogenic Genetic Diseases Based on Relative Haplotype Dosage Changes’. Genetics in Medicine Open, vol. 3, 2025, p. 103175. DOI.org (Crossref), https://doi.org/10.1016/j.gimo.2025.103175.
- Temaj, G., et al. ‘The Impact of Consanguinity on Human Health and Disease with an Emphasis on Rare Diseases’. Journal of Rare Diseases, vol. 1, no. 1, Dec. 2022, p. 2. DOI.org (Crossref), https://doi.org/10.1007/s44162-022-00004-5.
- Peterlin, Borut, and Ana Peterlin. ‘Carrier Screening and Pregnancy’. Best Practice & Research Clinical Obstetrics & Gynaecology, vol. 100, June 2025, p. 102601. DOI.org (Crossref), https://doi.org/10.1016/j.bpobgyn.2025.102601.
- Hotta, Yoshihiro, et al. ‘Ocular Genetics in the Japanese Population’. Japanese Journal of Ophthalmology, vol. 68, no. 5, Sept. 2024, pp. 401–18. DOI.org (Crossref), https://doi.org/10.1007/s10384-024-01109-8.
- Wang, Tianjiao, et al. ‘An Overview of Reproductive Carrier Screening Panels for Autosomal Recessive and/or X‐linked Conditions: How Much Do We Know?’ Prenatal Diagnosis, vol. 43, no. 11, Oct. 2023, pp. 1416–24. DOI.org (Crossref), https://doi.org/10.1002/pd.6434.
- Dive, Lisa, et al. ‘Ethical Considerations in Gene Selection for Reproductive Carrier Screening’. Human Genetics, vol. 141, no. 5, May 2022, pp. 1003–12. DOI.org (Crossref), https://doi.org/10.1007/s00439-021-02341-9.
- Edwards, Samantha, and Nigel Laing. ‘Genetic Counselling Needs for Reproductive Genetic Carrier Screening: A Scoping Review’. Journal of Personalized Medicine, vol. 12, no. 10, Oct. 2022, p. 1699. DOI.org (Crossref), https://doi.org/10.3390/jpm12101699.
- Prabhu, Akshatha. ‘Fetal Medicine and Current Practice of Prenatal Screening’. Apollo Medicine, vol. 20, no. 2, June 2023, pp. 135–38. DOI.org (Crossref), https://doi.org/10.4103/am.am_60_23.
- Veneruso, Iolanda, et al. ‘Current Updates on Expanded Carrier Screening: New Insights in the Omics Era’. Medicina, vol. 58, no. 3, Mar. 2022, p. 455. DOI.org (Crossref), https://doi.org/10.3390/medicina58030455.
- Srinivasan, Balaji S., et al. ‘A Universal Carrier Test for the Long Tail of Mendelian Disease’. Reproductive BioMedicine Online, vol. 21, no. 4, Oct. 2010, pp. 537–51. DOI.org (Crossref), https://doi.org/10.1016/j.rbmo.2010.05.012.
- Nguengang Wakap, Stéphanie, et al. ‘Estimating Cumulative Point Prevalence of Rare Diseases: Analysis of the Orphanet Database’. European Journal of Human Genetics, vol. 28, no. 2, Feb. 2020, pp. 165–73. www.nature.com, https://doi.org/10.1038/s41431-019-0508-0.
- Chung, Brian Hon Yin, et al. ‘Rare versus Common Diseases: A False Dichotomy in Precision Medicine’. Npj Genomic Medicine, vol. 6, no. 1, Feb. 2021, p. 19. DOI.org (Crossref), https://doi.org/10.1038/s41525-021-00176-x.
- Faye, Fatoumata, et al. ‘Time to Diagnosis and Determinants of Diagnostic Delays of People Living with a Rare Disease: Results of a Rare Barometer Retrospective Patient Survey’. European Journal of Human Genetics, vol. 32, no. 9, Sept. 2024, pp. 1116–26. DOI.org (Crossref), https://doi.org/10.1038/s41431-024-01604-z.
- Laing, Nigel G., et al. ‘Genetic Neuromuscular Disorders: What Is the Best That We Can Do?’ Neuromuscular Disorders, vol. 31, no. 10, Oct. 2021, pp. 1081–89. DOI.org (Crossref), https://doi.org/10.1016/j.nmd.2021.07.007.
- https://www.info.pmda.go.jp/downfiles/md/PDF/200880/200880_28B3X10006000050_A_01_01.pdf



