
Article Summary
Cri-du-chat syndrome is a rare genetic disorder characterized by a high-pitched, cat-like cry in infancy. It is caused by a partial deletion of the short arm of chromosome 5 (5p) and can lead to various physical, developmental, and behavioral challenges. Early diagnosis and comprehensive interventions can significantly improve developmental outcomes and quality of life.
Genes Associated With Disorders at the 17p13.1 Locus

The 17p13.1 locus includes multiple genes with known associations to rare disorders. These include:
- DLG4, associated with DLG4-related synaptopathy
- ACADVL, associated with very long-chain acyl-coenzyme A dehydrogenase deficiency
- CHRNB1, associated with congenital myasthenic syndrome
- MPDU1, associated with congenital disorders of N-linked glycosylation and multiple metabolic pathways
- TP53, associated with Wilms tumor predisposition and Li-Fraumeni syndrome
- WRAP53, associated with dyskeratosis congenita and related telomere biology disorders
- GUCY2D, associated with Leber congenital amaurosis and early-onset severe retinal dystrophy
- ALOX12B and ALOXE3, both associated with autosomal recessive congenital ichthyosis
- HES7, associated with congenital diaphragmatic hernia and spondylocostal dysostosis
- TMEM107, associated with Meckel syndrome 13 and Joubert syndrome
- CTC1, associated with dyskeratosis congenita and related telomere disorders
- RANGRF, associated with Brugada syndrome
- RPL26, associated with Diamond-Blackfan anemia
- NTN1, associated with congenital mirror movements
Each of these genes contributes to complex biological pathways, and alterations can produce a spectrum of phenotypes that vary in severity.
DLG4-Related Synaptopathy

Genetic Background
DLG4-related synaptopathy is a rare autosomal dominant genetic disorder caused by mutations in the DLG4 gene, which encodes the synaptic protein PSD-95 (postsynaptic density protein 95). PSD-95 is essential for regulating excitatory synapse function in the brain, where it plays a central role in synaptic stability and signaling.
Most cases result from de novo pathogenic variants, though familial transmission can occasionally occur through parental mosaicism or heterozygosity. The recurrence risk for families is generally considered low, but genetic counseling is recommended when a mutation is identified. Molecular confirmation through genetic testing allows for reproductive planning, including options such as prenatal or preimplantation genetic diagnosis.

Clinical Presentation
DLG4-related synaptopathy affects neurodevelopment and neurological function with variable severity. Core clinical features include:
- Developmental delay
- Intellectual disability, often mild to moderate in severity
- Autism spectrum disorder
About half of affected individuals experience epilepsy, and approximately 40 percent exhibit developmental regression, particularly affecting motor and language abilities.
Other neurological and systemic manifestations include:
- Hypotonia, or decreased muscle tone
- Movement disorders such as ataxia, dystonia, repetitive movements, and tremor
- Migraine headaches
- Sleep disturbances, including difficulty initiating and maintaining sleep
- Visual and ocular abnormalities such as strabismus, hyperopia, nystagmus, and cortical visual impairment
- Gastrointestinal symptoms, particularly vomiting triggered by seizures, motion sickness, or fatigue
- Connective tissue and musculoskeletal findings such as joint hypermobility (seen in about 37 percent of patients) and scoliosis (reported in around 20 percent)
Diagnosis
Diagnosis is established by molecular genetic testing that identifies a pathogenic variant in DLG4. Electroencephalography (EEG), including 24-hour monitoring, is recommended when seizures or developmental regression are suspected. Annual ophthalmologic evaluations are advised, with more frequent assessment when visual symptoms change.
Management
There is no curative treatment for DLG4-related synaptopathy. Management is symptom-focused and supportive, with an emphasis on improving quality of life. Care plans are typically multidisciplinary and may include:
- Developmental and educational support tailored to intellectual and behavioral needs
- Antiepileptic drugs for seizure control, chosen based on seizure type and individual response
- Interventions for motor dysfunction, including physical therapy for ataxia or dystonia
- Behavioral therapies for sleep disturbances, with medication considered for severe insomnia
- Ophthalmologic care to address strabismus, refractive errors, or other visual deficits
- Monitoring and support for behavioral challenges such as anxiety, attention-deficit hyperactivity disorder (ADHD), aggression, or self-injurious behaviors
Routine follow-up is essential. Neurological status, motor function, tone, and headache frequency should be reassessed periodically. As children age, evolving behavioral and developmental challenges may require additional therapeutic and educational adaptations.
Prognosis
Life expectancy is not well defined. Survival into adulthood has been documented, with one case reported at age 47, suggesting that long-term survival is possible with appropriate management and support. Earlier clinical descriptions highlighted features such as a Marfan-like body habitus or characteristic facial traits, but more recent studies show these findings are not consistently present. Disease severity is influenced primarily by the extent of synaptic dysfunction and resulting effects on neurodevelopment and behavior.
Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency

Genetic and Biochemical Background
Very long-chain acyl-coenzyme A dehydrogenase deficiency (VLCAD deficiency) is a rare inherited metabolic disorder caused by pathogenic variants in the ACADVL gene. This gene encodes the enzyme very long-chain acyl-CoA dehydrogenase, which is located in mitochondria and plays a key role in the breakdown of long-chain fatty acids during the beta-oxidation pathway. This pathway provides energy during fasting or periods of increased metabolic demand, such as illness or exercise.
When ACADVL function is impaired, the body cannot efficiently metabolize long-chain fatty acids. This leads to the accumulation of toxic intermediates and an energy deficit, particularly affecting organs with high energy requirements such as the heart, liver, and skeletal muscle.
Clinical Forms
VLCAD deficiency presents across a spectrum, with three major clinical forms classified according to age of onset and severity.
Early-Onset, Severe Form
The early-onset, severe form typically manifests within the first few months of life. It often presents with hypertrophic or dilated cardiomyopathy, pericardial effusion, arrhythmias, hypotonia, hepatomegaly, and hypoketotic hypoglycemia. Without rapid diagnosis and intervention, this form can lead to life-threatening metabolic crises and multi-organ failure.
Childhood-Onset, Hepatic Form
This intermediate form usually presents in early childhood with recurrent episodes of hypoketotic hypoglycemia and hepatomegaly, but without cardiomyopathy. These episodes are often triggered by prolonged fasting or illness.
Later-Onset, Myopathic Form
The late-onset, or myopathic, form often appears in adolescence or adulthood. It is characterized by exercise intolerance, muscle pain, cramps, rhabdomyolysis, and sometimes myoglobinuria, typically precipitated by exertion, fasting, or other metabolic stress. Hypoglycemia is rare in this form.
Diagnosis
The introduction of newborn screening (NBS) has significantly improved the early detection of VLCAD deficiency. Many infants identified through screening are asymptomatic at diagnosis, allowing for proactive management and prevention of severe complications. Laboratory studies typically show elevated acylcarnitines, and molecular genetic testing confirms the diagnosis by identifying pathogenic variants in ACADVL.
The estimated incidence of VLCAD deficiency is approximately 1 in 30,000 to 1 in 100,000 live births, though prevalence may vary by population.

Management
Management focuses on preventing metabolic crises and providing alternative energy sources. The cornerstone of treatment is dietary modification, which includes:
- Restricting long-chain fats
- Supplementing with medium-chain triglycerides (MCT), which bypass the defective enzyme pathway and serve as an alternative energy source
- Ensuring frequent meals and bedtime snacks, especially complex carbohydrates, to avoid fasting-induced hypoglycemia
In some cases, supplementation with carnitine, omega fatty acids, or vitamins A, D, and E may be recommended, though such use is individualized. Excessive physical exertion should be avoided to prevent muscle breakdown.
During acute metabolic crises, intravenous glucose and fluids are administered to stabilize blood glucose levels and prevent rhabdomyolysis. If rhabdomyolysis occurs, aggressive hydration and urine alkalinization may be needed to protect renal function. For patients undergoing surgery, careful perioperative management is critical to avoid metabolic decompensation.
Monitoring
Regular follow-up is essential. Monitoring includes growth and nutritional assessments, biochemical markers of metabolic control, cardiac evaluations such as echocardiograms, and assessments of skeletal muscle health.
Inheritance and Genetic Counseling
VLCAD deficiency follows an autosomal recessive inheritance pattern. When both parents are carriers of ACADVL pathogenic variants, each child has a 25 percent chance of being affected, a 50 percent chance of being a carrier, and a 25 percent chance of being unaffected. Genetic testing allows identification of carriers, enabling informed family planning and, if desired, prenatal or preimplantation genetic diagnosis.
Prognosis
With early diagnosis and careful management, even individuals with the severe early-onset form can achieve good long-term outcomes. Advances in screening, dietary therapy, and supportive care have markedly improved survival and quality of life. Lifelong monitoring and individualized management remain essential for all forms of VLCAD deficiency.
Congenital Myasthenic Syndrome

Genetic and Pathophysiological Background
Congenital myasthenic syndromes (CMS) are a group of rare genetic disorders affecting neuromuscular transmission. These disorders result from mutations in genes encoding proteins critical to the function of the neuromuscular junction. One of these genes is CHRNB1, which encodes the beta subunit of the acetylcholine receptor, a key protein mediating communication between nerve and muscle cells.
Mutations in CHRNB1 disrupt the transmission of signals from nerve endings to skeletal muscle fibers, leading to impaired muscle contraction and clinically significant weakness. Unlike autoimmune myasthenia gravis, CMS is not immune-mediated.
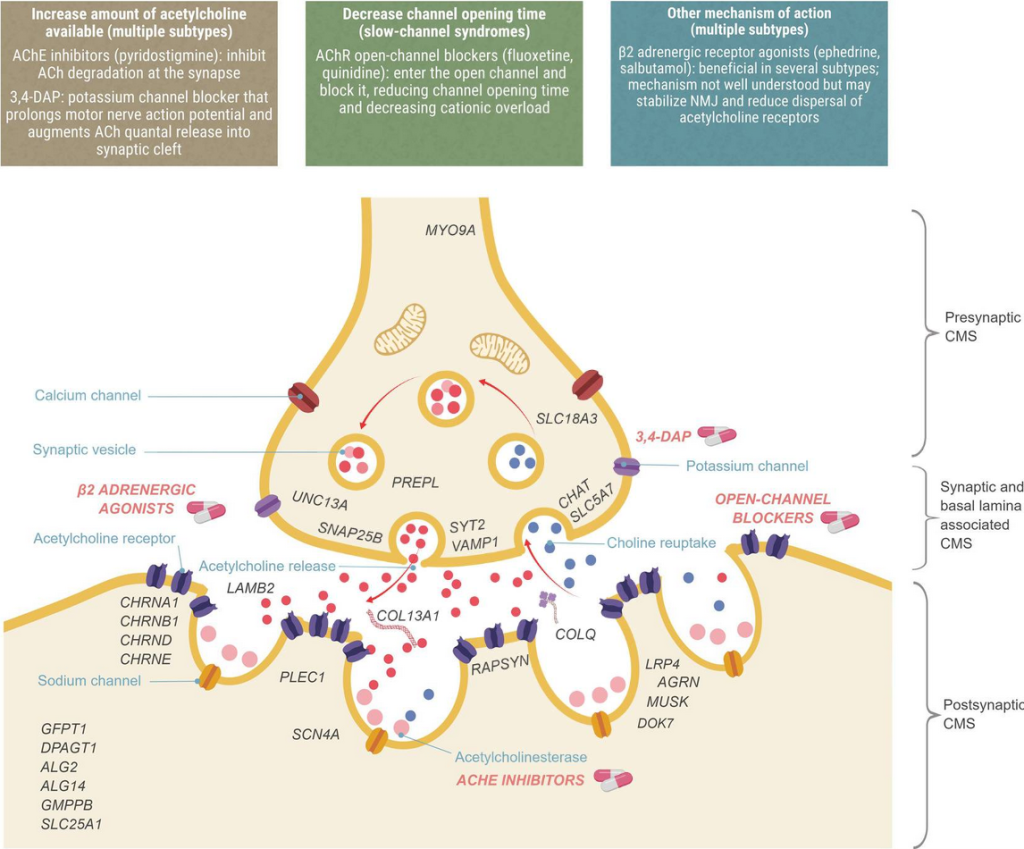
Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). [Figure 1. Localization of CMS types and therapeutic strategies.] Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
Clinical Presentation
CMS most often presents in the neonatal period or early infancy, typically within the first two years of life, though adult-onset cases have been reported. The severity and progression vary widely, depending on the specific subtype.
Common features include muscle weakness that worsens with activity and improves with rest. In neonates, clinical signs may include intermittent apnea, cyanosis, poor feeding, weak suck and cry, and generalized hypotonia. Some infants also display decreased fetal movements during pregnancy, which may lead to congenital contractures such as arthrogryposis.
During infancy and childhood, affected individuals may exhibit delayed motor milestones, eyelid drooping (ptosis), weakness of the eye muscles, facial muscle weakness, and difficulties with speech or swallowing. Some develop a limb-girdle pattern of weakness, affecting the shoulders and hips, which can interfere with walking and stair climbing. These symptoms may worsen during infections, stress, or fever.
Cognition, coordination, sensation, and reflexes are typically normal, although a subset of subtypes is associated with more complex neurological presentations.
Diagnosis
Diagnosis relies on clinical assessment, neurophysiological studies, and confirmation through genetic testing. Identifying the specific molecular defect is critical because different subtypes respond to different treatments. Brain imaging is typically normal in CHRNB1-associated CMS, distinguishing it from other neuromuscular disorders.
Management
Treatment is highly individualized and depends on the CMS subtype. For CHRNB1-associated CMS, the following medications are commonly used:
- Pyridostigmine, an acetylcholinesterase inhibitor that enhances signal transmission at the neuromuscular junction
- Fluoxetine and quinidine, often considered first-line options alongside pyridostigmine
- Salbutamol or ephedrine as adjunctive therapies for additional symptom control
Other subtypes may respond to agents such as 3,4-diaminopyridine (3,4-DAP), but these are not universally effective and can exacerbate symptoms in some variants. Close medical supervision is required to monitor response and side effects.
Supportive and Multidisciplinary Care
Non-pharmacological interventions are also important. Physical and occupational therapy can help maintain mobility and prevent contractures. Speech and feeding therapy support safe swallowing and communication skills. In severe cases, wheelchairs, orthotics, or ventilatory support may be required. Gastrostomy tubes may be indicated for individuals with significant feeding difficulties.
For infants with apnea, home apnea monitors and parental training in basic life support are recommended. During pregnancy, women with CMS require close respiratory and cardiac monitoring under specialist care.
Monitoring and Prognosis
Regular follow-up is crucial, with evaluations recommended every six months for children and annually for adults. Monitoring includes assessments of muscle strength, respiratory function, and overall health. While CMS is a lifelong condition, symptoms can often be effectively managed with appropriate therapy, allowing many individuals to achieve a good quality of life.
MPDU1-Related Congenital Disorders of Glycosylation

Genetic and Molecular Background
The MPDU1 gene encodes a protein essential for the proper assembly of dolichol-linked oligosaccharides, which are critical for N-linked glycosylation and multiple metabolic pathways. This biochemical process ensures that glycoproteins are correctly folded and functional, influencing everything from cell signaling to structural integrity.Mutations in MPDU1 disrupt this pathway, leading to congenital disorders of glycosylation (CDG). These disorders are rare, typically inherited in an autosomal recessive pattern, and exhibit considerable clinical variability depending on the extent of disruption in glycosylation.
Clinical Presentation
Infants with MPDU1-related CDG often present with a constellation of systemic and neurological symptoms. Common early findings include:
- Severe developmental delay and hypotonia
- Failure to thrive due to feeding difficulties
- Seizures that can range from manageable to refractory
- Dysmorphic facial features, which may include a prominent forehead, thin upper lip, or abnormal ear morphology
Systemic complications often include hepatomegaly and coagulopathy, sometimes progressing to multi-organ involvement. Cardiomyopathy has been reported in severe cases. Neurological features are prominent and may include microcephaly, movement disorders, and profound intellectual disability.
Diagnosis
Diagnosis is suspected based on clinical features combined with laboratory evidence of abnormal glycosylation patterns, usually detected via serum transferrin isoelectric focusing or mass spectrometry. Genetic testing of the MPDU1 gene confirms the diagnosis and enables family counseling.
Because glycosylation defects often impact multiple organ systems, a multidisciplinary assessment is recommended during the diagnostic phase, including neurology, gastroenterology, and cardiology evaluations.

Management
There is currently no curative therapy for MPDU1-related CDG. Management is supportive and symptom-based, focusing on improving quality of life and preventing complications. This often includes:
- Anti-seizure medications tailored to the seizure profile
- Nutritional support, including gastrostomy tube feeding for severe feeding difficulties
- Physical, occupational, and speech therapy to maximize developmental outcomes
- Surveillance for liver dysfunction, coagulopathy, and cardiac complications
Because the disease course is often progressive, early palliative care involvement may be appropriate for families managing severe phenotypes.

Prognosis
The prognosis depends on the severity of the enzyme deficiency and the degree of systemic involvement. Severe cases often lead to early mortality, while milder cases may survive into childhood or adolescence with persistent neurodevelopmental impairment. Genetic counseling is critical for affected families, as recurrence risk is 25 percent for each pregnancy when both parents are carriers.
TP53-Associated Tumor Predisposition Syndromes

Li-Fraumeni Syndrome
Li-Fraumeni syndrome is characterized by early-onset and multiple primary cancers across a broad spectrum. The most common malignancies include:
- Soft tissue sarcomas
- Osteosarcomas
- Pre-menopausal breast cancer
- Brain tumors
- Adrenocortical carcinoma
- Leukemias
Affected individuals often develop cancer before the age of 30, and many experience two or more distinct malignancies during their lifetime. Penetrance is high, with lifetime cancer risk approaching 70 percent in men and over 90 percent in women.
De novo mutations account for approximately 7 to 20 percent of LFS cases, meaning some patients have no family history of the syndrome.
Wilms Tumor Predisposition
Mutations in TP53 also contribute to a smaller subset of hereditary Wilms tumor cases, though this presentation is less common than the classic Li-Fraumeni phenotype.
Diagnosis
Diagnosis is established through molecular genetic testing of TP53. Clinical criteria, such as the Chompret criteria, are often used to identify individuals for testing, particularly those with early-onset cancers or family histories suggestive of LFS.

Management and Surveillance
There is no preventive therapy to reverse the mutation’s effect, so management is focused on early detection and risk reduction. Recommended surveillance strategies include:
- Annual whole-body MRI to screen for early malignancies without radiation exposure
- Brain MRI every one to three years
- Dermatologic, breast, and abdominal imaging based on age and risk factors
- Avoidance of radiation therapy when possible, due to heightened radiosensitivity in TP53 mutation carriers
For female carriers, breast MRI is recommended starting in early adulthood, with risk-reducing mastectomy considered on an individual basis.

Family Planning and Counseling
Genetic counseling is essential. Each child of an affected parent has a 50 percent chance of inheriting the mutation. Preimplantation or prenatal genetic diagnosis may be considered for family planning.
Prognosis
The prognosis for TP53-associated cancer predisposition depends heavily on the type and stage of cancers that develop, as well as the effectiveness of surveillance and early detection. Advances in comprehensive screening protocols have improved survival outcomes for affected individuals by enabling earlier diagnosis and intervention.
WRAP53-Related Dyskeratosis Congenita and Telomere Biology Disorders

Genetic and Molecular Basis
The WRAP53 gene encodes a protein that is essential for the maintenance of telomere integrity and stability. Telomeres, the protective caps at the ends of chromosomes, are critical for genomic stability and cell division. Mutations in WRAP53 lead to defective telomere maintenance, resulting in accelerated telomere shortening and impaired cellular repair mechanisms.
Pathogenic variants in this gene are linked to dyskeratosis congenita (DC) and related telomere biology disorders, which exhibit a wide spectrum of severity depending on the extent of telomere dysfunction.
Clinical Features
Dyskeratosis congenita often presents with a classic triad:
- Abnormal skin pigmentation
- Nail dystrophy
- Oral leukoplakia
However, clinical manifestations are variable and may extend to other systems, reflecting the multi-tissue consequences of telomere dysfunction. Additional findings can include:
- Bone marrow failure, leading to cytopenias
- Pulmonary fibrosis and progressive respiratory decline
- Liver disease, sometimes progressing to cirrhosis
- Increased predisposition to malignancies, including leukemias and squamous cell carcinomas
Age of onset ranges widely, from early childhood in severe cases to adulthood in milder phenotypes. Severe presentations, such as Hoyeraal-Hreidarsson syndrome or Revesz syndrome, are typically seen in individuals with critically short telomeres and present with early-onset neurological impairment, intrauterine growth restriction, and severe immunodeficiency.
Diagnosis
Diagnosis combines clinical evaluation, telomere length measurement, and genetic confirmation of WRAP53 mutations. Telomere length analysis via flow cytometry and fluorescent in situ hybridization (flow-FISH) is a standard laboratory test. Molecular genetic testing confirms the pathogenic variant and guides family counseling.
Management
There is no curative therapy for WRAP53-related telomere biology disorders. Management focuses on surveillance, prevention, and treatment of complications. Key approaches include:
- Hematologic monitoring with regular blood counts to detect early bone marrow failure
- Androgen therapy or hematopoietic stem cell transplantation (HSCT) for progressive bone marrow failure
- Pulmonary monitoring, including lung function tests and imaging, to detect early pulmonary fibrosis
- Cancer surveillance, given the significantly increased lifetime risk for malignancies
Supportive care also addresses oral leukoplakia, dermatologic symptoms, and management of liver disease.
Patients undergoing HSCT require special consideration, as their underlying telomere dysfunction increases the risk of transplant-related complications.

Prognosis
Prognosis varies by phenotype and degree of telomere shortening. Severe early-onset cases often result in significant morbidity and early mortality, primarily from bone marrow failure or pulmonary complications. Individuals with milder variants may live into adulthood but remain at risk for cancer and organ failure. Genetic counseling is essential to discuss inheritance, which is typically autosomal recessive, though dominant patterns have been documented in some families.
GUCY2D-Associated Retinal Dystrophies

Genetic Background and Pathogenesis
The GUCY2D gene encodes retinal guanylate cyclase 1, an enzyme crucial for phototransduction in photoreceptor cells. Pathogenic variants in GUCY2D lead to two main clinical conditions:
- Leber Congenital Amaurosis (LCA)
- Early-Onset Severe Retinal Dystrophy (EOSRD)
Both disorders arise from disrupted visual signal transduction, resulting in severe visual impairment from birth or early infancy. The inheritance pattern is typically autosomal recessive, although autosomal dominant variants have been reported in specific contexts.
Clinical Presentation
In Leber congenital amaurosis, affected infants often present in the first year of life with:
- Severe visual impairment or blindness
- Nystagmus, often noticed by caregivers within months of birth
- Poor pupillary responses
- Eye-poking or pressing behaviors (oculodigital sign), which may lead to enophthalmos over time
Early-onset severe retinal dystrophy manifests similarly but may allow for slightly better initial visual function, with progressive vision loss over early childhood. Fundus examination often shows retinal pigmentary changes, and electroretinography (ERG) demonstrates severely reduced or absent retinal responses.
Other systemic features are uncommon, distinguishing these conditions from syndromic retinal dystrophies such as those caused by CEP290 or CRB1 variants.
Diagnosis
Diagnosis is based on a combination of clinical findings, ERG testing, and molecular genetic testing to confirm pathogenic GUCY2D variants. Optical coherence tomography (OCT) imaging often reveals preserved retinal architecture despite profound functional impairment, an observation that has driven interest in gene replacement therapies.
Management
Currently, management is primarily supportive, focusing on optimizing residual vision and providing adaptive resources. This includes:
- Early referral to low-vision services
- Use of high-contrast materials and adaptive educational strategies
- Orientation and mobility training for older children
- Genetic counseling for families
Experimental treatments, including gene therapy approaches such as subretinal delivery of functional GUCY2D, are in advanced stages of clinical trials, offering future therapeutic potential.

Prognosis
Vision is typically severely impaired from early life and often remains stable, although some individuals experience progressive deterioration over time. Quality of life can be significantly improved through early educational intervention and adaptive support systems. Genetic counseling is essential for family planning, given the autosomal recessive inheritance pattern and 25 percent recurrence risk for siblings.
ALOX12B and ALOXE3-Related Autosomal Recessive Congenital Ichthyosis


Genetic and Molecular Basis
Autosomal recessive congenital ichthyosis (ARCI) encompasses a group of rare keratinization disorders caused by pathogenic variants in genes critical for skin barrier formation. Among these, ALOX12B and ALOXE3 encode lipoxygenases that metabolize epidermal lipids, supporting the integrity and function of the stratum corneum. Mutations in these genes impair the formation of the skin barrier, resulting in excessive scaling and hyperkeratosis from birth.
Both conditions are inherited in an autosomal recessive pattern. Parents of an affected child are typically asymptomatic carriers, with a 25 percent recurrence risk in subsequent pregnancies.
Clinical Presentation
Affected newborns frequently present as collodion babies, encased in a shiny, taut membrane that may peel off over the first weeks of life. As the membrane sheds, it reveals varying degrees of ichthyosis, ranging from fine scaling to thick, plate-like hyperkeratosis. Associated findings may include:
- Ectropion (outward turning of the eyelids)
- Eclabium (outward turning of the lips)
- Flexion contractures of fingers or toes due to skin tightening
- Increased risk of dehydration and infection in the neonatal period
Over time, affected individuals often display generalized scaling with varying severity, often accompanied by erythema. The condition tends to persist throughout life, although severity and pattern of scaling may evolve with age.
Pruritus and heat intolerance are common, as impaired sweating disrupts thermoregulation. Hair and nail involvement is less common but can occur in severe phenotypes.
Diagnosis
Diagnosis is based on clinical presentation and family history, supported by molecular genetic testing to confirm pathogenic variants in ALOX12B or ALOXE3. Histopathological studies are rarely required but may show nonspecific hyperkeratosis.

Management
There is no curative therapy for ARCI. Management focuses on supportive skin care and prevention of complications. This includes:
- Frequent application of emollients and keratolytic creams to reduce scaling and maintain skin hydration
- Humidified environments to minimize transepidermal water loss
- Topical retinoids or, in severe adolescent or adult cases, systemic retinoids such as acitretin under careful medical supervision
- Surveillance for secondary skin infections, particularly in the neonatal period
Supportive care in neonates is critical, with close monitoring for dehydration, infection, and respiratory compromise due to skin tightening.

Prognosis
With appropriate skin care, individuals with ALOX12B- or ALOXE3-related ichthyosis typically achieve normal growth and development. However, the condition remains lifelong and may have psychosocial implications, particularly in visible or severe cases. Genetic counseling is recommended for families regarding recurrence risk and reproductive options.
HES7-Associated Disorders

Genetic and Molecular Background
The HES7 gene encodes a transcription factor involved in somite segmentation during embryonic development. Mutations in HES7 disrupt normal somitogenesis, leading to congenital skeletal and organ malformations. These mutations are associated with several developmental disorders, most notably:
- Spondylocostal Dysostosis (SCD)
- Congenital Diaphragmatic Hernia (CDH)
Both conditions are inherited in an autosomal recessive pattern when caused by biallelic loss-of-function mutations, although heterozygous variants have been associated with milder phenotypes.

Spondylocostal Dysostosis
Clinical Features
Spondylocostal dysostosis is characterized by multiple vertebral segmentation defects and rib anomalies. Clinical findings often include:
- Short trunk due to shortened thoracic spine
- Scoliosis, often progressive during childhood
- Restrictive lung disease due to thoracic cage deformity
- Short stature in severe cases
Despite skeletal anomalies, cognitive development is usually normal, though respiratory complications may impact quality of life.
Diagnosis
Diagnosis is typically established with spine and rib imaging revealing vertebral segmentation defects and rib anomalies. Molecular confirmation through genetic testing identifies biallelic HES7 variants, enabling accurate recurrence risk counseling for families.
Management
Management focuses on respiratory support and orthopedic interventions. For mild deformities, observation and physical therapy may suffice. In progressive scoliosis or severe thoracic restriction, surgical interventions such as growing rods or spinal fusion may be considered. Multidisciplinary care involving pulmonologists and orthopedic surgeons is essential.

Congenital Diaphragmatic Hernia
Clinical Features
Pathogenic HES7 variants have also been identified in cases of congenital diaphragmatic hernia (CDH), a structural defect of the diaphragm that allows abdominal organs to herniate into the thoracic cavity. Clinical presentation typically includes:
- Respiratory distress at birth
- Hypoplastic lungs
- Pulmonary hypertension
The severity of CDH varies widely, ranging from mild defects that require limited intervention to severe cases associated with high neonatal mortality.
Diagnosis and Management
Prenatal ultrasound often detects CDH. After birth, diagnosis is confirmed by imaging, typically chest radiography. Management includes stabilization of respiratory function, often with mechanical ventilation or extracorporeal membrane oxygenation (ECMO), followed by surgical repair. Survivors require long-term follow-up for respiratory and nutritional complications.
Prognosis
For spondylocostal dysostosis, prognosis depends on the severity of spinal and thoracic deformities. With appropriate management, many individuals reach adulthood with preserved function. For congenital diaphragmatic hernia, survival has improved due to advances in neonatal care and surgical techniques, though long-term complications such as pulmonary morbidity and gastroesophageal reflux are common.
TMEM107-Related Ciliopathies

Genetic and Cellular Basis
TMEM107 encodes a transmembrane protein that localizes to the ciliary transition zone. Primary cilia act as cellular antennae that gate key developmental signals, including Sonic hedgehog, WNT, and PDGF pathways. Pathogenic TMEM107 variants disrupt transition zone architecture and alter ciliary number and morphology, which in turn deranges signal transduction during organogenesis. The result is a ciliopathy spectrum with phenotypes that range from lethal malformations to survivable neurodevelopmental disorders. Biallelic loss of function typically produces severe disease. Hypomorphic or missense variants can yield milder phenotypes.
Clinical Spectrum: Meckel Syndrome 13
Meckel syndrome 13 is a severe, usually lethal ciliopathy inherited in an autosomal recessive pattern. Hallmark features include cystic renal disease, encephalocele arising from the occipital region, and congenital hepatic fibrosis with ductal plate malformation. Many fetuses also have postaxial polydactyly and posterior fossa abnormalities such as cerebellar vermis hypoplasia. Most pregnancies end in perinatal death because of the combination of central nervous system malformations, pulmonary hypoplasia, and multiorgan involvement.
Clinical Spectrum: Joubert Syndrome Associated With TMEM107
Joubert syndrome presents with the molar tooth sign on brain MRI, reflecting cerebellar vermis hypoplasia and brainstem malformation. Infants commonly show developmental delay, generalized hypotonia, and episodic neonatal breathing irregularities. Ocular motor apraxia and variable visual impairment can occur. Some individuals have systemic features that overlap with other ciliopathies, including polydactyly, hepatic fibrosis, and renal involvement. Within families, TMEM107 variants can produce Meckel syndrome in one pregnancy and Joubert syndrome in another, illustrating a shared genetic basis with graded phenotypic severity.

Diagnosis
Prenatal ultrasound and fetal MRI can detect encephalocele, enlarged echogenic kidneys, or posterior fossa anomalies. Postnatal evaluation relies on clinical examination, MRI for the molar tooth sign when Joubert syndrome is suspected, and genetic testing that identifies biallelic TMEM107 variants. When a familial variant is known, targeted testing enables early diagnosis. For at-risk pregnancies, cell-free DNA screening cannot diagnose specific ciliopathies but can guide decisions about definitive diagnostic testing through chorionic villus sampling or amniocentesis.
Management
There is no curative therapy. Care is supportive and organ directed. For Meckel syndrome, counseling focuses on prognosis, palliative planning, and discussion of future reproductive options. For Joubert syndrome, coordinated care by neurology, nephrology, hepatology, ophthalmology, and developmental services is central. Early intervention, physical and occupational therapy, and vision support improve function. Surgical management may be indicated for polydactyly or cleft palate when present. Nutrition, growth, and respiratory status require routine monitoring in infancy.
Prognosis and Counseling
Meckel syndrome has a poor prognosis with high perinatal mortality. Joubert syndrome has wide variability, with many patients surviving into childhood and adulthood with ongoing developmental needs. TMEM107-related disease is autosomal recessive. Each sibling of an affected individual has a 25 percent risk of being affected, a 50 percent risk of being an asymptomatic carrier, and a 25 percent chance of inheriting neither pathogenic variant. When familial variants are known, options include prenatal diagnosis and preimplantation genetic testing.
CTC1-Related Coats Plus Syndrome and Telomere Biology Disorders

Genetic and Molecular Basis
CTC1 encodes a core subunit of the CST complex, a heterotrimer that safeguards telomere replication. CST binds single-stranded telomeric DNA, coordinates termination of telomerase activity, and promotes fill-in synthesis of the C-rich strand. Pathogenic CTC1 variants impair telomere replication and genome stability. The resulting disorder is classically described as Coats plus syndrome, also called cerebroretinal microangiopathy with calcifications and cysts type 1.

Clinical Features
The phenotype spans neurological, ocular, skeletal, hematologic, hepatic, and gastrointestinal systems. In the central nervous system, characteristic findings include intracranial calcifications, white matter injury, and parenchymal cysts, which together can produce spasticity, ataxia, dystonia, seizures, and cognitive impairment. In the retina, telangiectatic and aneurysmal vessels with lipid exudation produce a Coats-like retinopathy that threatens vision. Skeletal fragility with osteopenia or osteoporosis increases fracture risk. Gastrointestinal vascular malformations can lead to bleeding and portal hypertension. Cytopenias and nail, hair, and skin changes may reflect broader telomere biology dysfunction. The disorder is typically autosomal recessive, with biallelic CTC1 variants identified in affected individuals.
Diagnosis
Neuroimaging demonstrates calcifications, white matter changes, and cysts. Ophthalmologic examination confirms exudative retinopathy. Laboratory evaluation may show cytopenias and hepatic dysfunction. Molecular confirmation rests on identifying pathogenic variants in CTC1. The differential diagnosis includes Labrune syndrome, which shares brain calcifications and cysts but lacks the consistent systemic features, and other telomere disorders that present with bone marrow failure and pulmonary complications.
Management
Care is multidisciplinary and supportive. Ophthalmologic therapies such as laser photocoagulation or intravitreal agents can manage retinal exudation in selected cases. Neurological complications are addressed with antiepileptic medications, spasticity management, and rehabilitative therapies. Bone health requires calcium and vitamin D optimization, weight-bearing activity as tolerated, and fracture prevention strategies. Gastrointestinal bleeding and portal hypertension call for coordinated care with gastroenterology and interventional radiology. Hematology input is important for cytopenias and perioperative planning. Because the telomere defect increases treatment toxicity, any consideration of hematopoietic stem cell transplantation or cytotoxic therapy requires careful risk assessment.
Prognosis and Counseling
Disease course is variable and often progressive, driven by neurological decline, retinal disease, skeletal fragility, or gastrointestinal complications. Survival into adulthood is possible, particularly with vigilant surveillance and timely organ-specific interventions. Inheritance is autosomal recessive. Recurrence risk for carrier parents is 25 percent per pregnancy. When familial variants are known, prenatal diagnosis and preimplantation genetic testing are available. Families benefit from coordinated genetic counseling that also reviews overlapping features with other telomere biology disorders and outlines anticipatory screening for complications.
RANGRF-Related Brugada Syndrome

Genetic and Electrophysiological Basis
RANGRF encodes MOG1, a trafficking cofactor that helps deliver the cardiac sodium channel Nav1.5 to the myocyte membrane. Variants in RANGRF can reduce membrane localization of Nav1.5, lowering peak sodium current and predisposing to conduction abnormalities that underlie Brugada syndrome. Brugada syndrome is genetically heterogeneous. SCN5A variants are the most common cause, while RANGRF and several other genes contribute a smaller proportion of cases. The disorder is usually inherited in an autosomal dominant pattern with reduced penetrance.
Clinical Features
Typical presentation is in adulthood, although cases are reported from infancy to older age. The average age for sudden death is about 40 years. Men are affected far more often than women, with an approximate 8 to 1 ratio. Hallmark findings include a characteristic electrocardiographic pattern in the right precordial leads V1 to V3 with coved ST segment elevation and a propensity for ventricular arrhythmias. Symptoms range from none to syncope or cardiac arrest. Brugada syndrome has been implicated in a fraction of sudden infant death cases and is linked to sudden unexpected nocturnal death syndrome described in Southeast Asia.
Diagnosis
Diagnosis rests on the type 1 Brugada ECG pattern, either spontaneous or unmasked by a sodium channel blocker challenge performed under specialist supervision. Clinical history and family history refine pretest probability. Genetic testing can identify pathogenic variants in SCN5A or other Brugada-associated genes including RANGRF, which helps with cascade testing in relatives but is not required for diagnosis.
Management
The primary goal is prevention of sudden death. An implantable cardioverter defibrillator is indicated for survivors of cardiac arrest and for individuals with syncope attributable to ventricular arrhythmia. Quinidine can reduce arrhythmia burden in selected high risk patients and is sometimes used to reduce implantable cardioverter defibrillator shocks. Catheter ablation targeting the arrhythmogenic substrate of the right ventricular outflow tract epicardium is an option in recurrent ventricular fibrillation. All patients should avoid fever when possible, promptly treat febrile illness, and avoid drugs known to exacerbate Brugada phenotype. Regular follow up with ECG review is advisable in genetically at risk relatives even if asymptomatic.

Inheritance, Epidemiology, and Prognosis
Inheritance is usually autosomal dominant with variable expressivity. Some cases arise from de novo variants. Global prevalence is estimated at approximately 1 in 2,000, with higher prevalence in parts of Asia and the Middle East. Many individuals remain asymptomatic. Among symptomatic cohorts, about 30 percent report syncope and 8 to 12 percent experience cardiac arrest. Prognosis improves with risk stratification, fever avoidance, appropriate device therapy when indicated, and family screening.
RPL26-Related Diamond-Blackfan Anemia

Molecular Pathogenesis
RPL26 encodes a component of the 60S ribosomal subunit. Pathogenic variants disrupt ribosome biogenesis and impair erythropoiesis, producing Diamond-Blackfan anemia type 11. Diamond-Blackfan anemia is a ribosomopathy that primarily reduces red blood cell production while sparing other hematopoietic lineages, although multilineage effects and systemic anomalies can occur.

Clinical Features
The cardinal presentation is moderate to severe macrocytic anemia within the first year of life, accompanied by low reticulocyte counts and a paucity of erythroid precursors in the bone marrow. White blood cell and platelet counts are often normal. About 30 to 40 percent of patients have congenital anomalies that may include craniofacial differences such as cleft palate or Pierre Robin sequence, thumb or radial ray malformations, and urogenital anomalies. Severe fetal anemia can cause hydrops in rare cases. There is an increased lifetime risk of malignancy, including acute myeloid leukemia, myelodysplastic syndrome, and osteosarcoma.
Diagnosis
Diagnosis combines clinical timing and morphology of anemia, reticulocytopenia, and marrow findings with exclusion of secondary causes. Molecular confirmation is achieved by identifying a pathogenic variant in an established Diamond-Blackfan anemia gene such as RPL26. Most cases follow autosomal dominant inheritance with variable penetrance. X-linked forms due to GATA1 or TSR2 have been described.
Management
Therapeutic priorities are correction of anemia and prevention of complications. Corticosteroids after 12 months of age can increase red blood cell counts in many patients, with careful monitoring for adverse effects such as infection risk, growth suppression, and bone demineralization. If steroid response is inadequate or toxicities are unacceptable, chronic red blood cell transfusions are used with iron chelation to prevent iron overload. Hematopoietic stem cell transplantation is the only curative option for the hematologic phenotype and is considered for transfusion dependent or refractory disease. Multidisciplinary care addresses skeletal, ophthalmologic, endocrine, and other associated anomalies. Oncology teams should tailor chemotherapy cautiously if cancer arises due to baseline marrow vulnerability.

Monitoring, Genetics, and Pregnancy Considerations
Surveillance includes periodic complete blood counts, iron studies to guide chelation, bone health evaluation, and age appropriate cancer screening. Family members can be tested for the familial variant to clarify recurrence risk and permit early intervention. During pregnancy, close monitoring of maternal hemoglobin is recommended. Low dose aspirin may be used in selected cases to mitigate obstetric risks, coordinated by maternal fetal medicine and hematology.
NTN1-Related Congenital Mirror Movements

Developmental Neurobiology
NTN1 encodes netrin 1, a guidance cue that directs commissural and corticospinal axons through attraction and repulsion signals mediated by receptors such as DCC and members of the UNC5 family. Netrin 1 also supports neuronal survival by modulating apoptosis pathways. Variants that reduce netrin 1 function can disrupt the decussation of descending motor pathways, leading to persistent mirror movements.

Clinical Features
Congenital mirror movements are involuntary movements in one limb that mirror voluntary movements of the opposite limb. Onset is in early childhood. Movements are most evident in the upper limbs, especially during fine motor tasks of the hands and fingers. Gait is usually normal, although mild toe involvement can occur. The condition is nonprogressive and varies in severity. Pain or fatigue of the upper limbs can occur with repetitive tasks. Cognition and brain imaging are typically normal in NTN1-related cases. By contrast, DCC-related mirror movements can be associated with partial or complete agenesis of the corpus callosum, a finding not reported to date in NTN1-related disease.
Diagnosis
Diagnosis is clinical, supported by family history and confirmed by molecular testing that identifies a heterozygous pathogenic variant in NTN1. Electrophysiology can demonstrate abnormal bilateral motor activation during unilateral tasks. Brain MRI is usually normal in NTN1-related cases. The differential diagnosis includes physiologic mirror movements that resolve by about age seven, movement disorders, pyramidal tract disorders, and functional neurologic conditions.

Management
There is no disease modifying therapy. Management focuses on adaptation rather than elimination of movements. Educational accommodations such as extended time for writing tasks, reduced handwriting load, or use of keyboards can improve performance in school. Occupational therapy targets task strategies that minimize bimanual interference. In adolescence and adulthood, career choices that avoid prolonged fine bimanual work may reduce discomfort. Psychological support can mitigate social stigma and reinforce strengths.
Inheritance, Epidemiology, and Outlook
NTN1-related mirror movements follow autosomal dominant inheritance with reduced penetrance. A parent can carry the variant without overt symptoms, and asymptomatic transmission to relatives is possible. Estimated prevalence is lower than 1 per 1,000,000, but true frequency is likely higher due to underdiagnosis of mild cases. The condition is lifelong yet stable, and most individuals adapt effectively with targeted supports.
Prenatal Screening Context and Family Planning
Role of NIPT and Diagnostic Testing
Noninvasive prenatal testing analyzes cell free fetal DNA in maternal plasma. It is a screening test that estimates risk for common aneuploidies and some copy number changes. It does not diagnose single gene disorders such as those described here. When a familial pathogenic variant is known, prenatal diagnosis requires chorionic villus sampling or amniocentesis for targeted molecular testing. Preimplantation genetic testing is an alternative for some families planning in vitro fertilization.
Counseling Principles
For autosomal recessive conditions, carrier parents face a 25 percent recurrence risk in each pregnancy. For autosomal dominant conditions, each child of an affected individual has a 50 percent chance of inheriting the variant, with penetrance and expressivity guiding individualized risk discussions. Genetic counseling supports informed choices, clarifies surveillance plans, and connects families with clinical trials or registries when available.
Summary of the Full Article
The 17p13.1 chromosomal region carries many genes essential for healthy brain development, metabolism, telomere maintenance, and organ structure. When changes occur in these genes, they can lead to a wide range of rare disorders with different levels of severity.
Some conditions, such as DLG4-related synaptopathy, VLCAD deficiency, and congenital myasthenic syndrome, often show good outcomes with early diagnosis and appropriate management. Others, like telomere biology disorders, GUCY2D-related retinal diseases, or ciliopathies, currently lack curative treatments, but advances in science have improved understanding, allowing for better surveillance, early interventions, and informed family planning.
For families, a key source of hope is the growing precision of molecular testing. These tools provide clarity in diagnosis, help predict inheritance risks, and open the door to prenatal or preimplantation genetic testing when desired.
For individuals living with these conditions, multidisciplinary care and close monitoring can significantly improve quality of life. Children with developmental delays or metabolic instability often show steady progress with tailored therapies. Adults with metabolic or cardiac disorders are experiencing longer, more stable lives through modern interventions, such as dietary therapy in VLCAD deficiency or close cardiac surveillance in Brugada syndrome. Even for those with more complex or severe presentations, supportive care is increasingly effective at improving comfort, function, and connection.
Research continues to bring new opportunities. Gene therapy trials for GUCY2D-related vision loss are underway. Newborn screening programs are identifying metabolic conditions earlier than ever before, allowing for proactive management. Collaborative studies are refining care strategies across the spectrum of disorders linked to the 17p13.1 region.
Daily life with these diagnoses can be challenging, but knowledge, early action, and connection with experienced care teams are powerful. Families are finding new pathways toward stability, growth, and, in many cases, better long-term outcomes.

References
- Méneret A, Trouillard O, Dunoyer M, et al. Congenital Mirror Movements. 2015 Mar 12 [Updated 2020 Sep 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279760/
- Janov, A. J., Leong, T., Nathan, D. G., & Guinan, E. C. (1996). Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine, 75(2), 77–78. https://doi.org/10.1097/00005792-199603000-00004
- Willig, T.-N., Niemeyer, C. M., Leblanc, T., Tiemann, C., Robert, A., Budde, J., Lambiliotte, A., Kohne, E., Souillet, G., Eber, S., Stephan, J.-L., Girot, R., Bordigoni, P., Cornu, G., Blanche, S., Guillard, J. M., & Mohandas, N. (1999). Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 diamond-blackfan anemia patients. Pediatric Research, 46(5), 553–553. https://doi.org/10.1203/00006450-199911000-00011
- Sieff C. Diamond-Blackfan Anemia. 2009 Jun 25 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7047/
- Marfatia, K. A., Harreman, M. T., Fanara, P., Vertino, P. M., & Corbett, A. H. (2001). Identification and characterization of the human MOG1 gene. Gene, 266(1-2), 45–56. https://doi.org/10.1016/s0378-1119(01)00364-x
- Brugada R, Campuzano O, Sarquella-Brugada G, et al. Brugada Syndrome. 2005 Mar 31 [Updated 2022 Aug 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1517/
- Campuzano, O., Berne, P., Selga, E., Allegue, C., Iglesias, A., Brugada, J., & Brugada, R. (2014). Brugada syndrome and p.E61X_RANGRF. Cardiology journal, 21(2), 121–127. https://doi.org/10.5603/CJ.a2013.0125
- Kayarian, F. B., Cohen, S. M., Cohen, M. L., & Sammartino, D. E. (2023). Coats plus syndrome presenting in an adult. Journal of VitreoRetinal Diseases, 7(6), 562–564. https://doi.org/10.1177/24741264231171465
- Anderson, B. H., Kasher, P. R., Mayer, J., Szynkiewicz, M., Jenkinson, E. M., Bhaskar, S. S., Urquhart, J. E., Daly, S. B., Dickerson, J. E., O’Sullivan, J., Leibundgut, E. O., Muter, J., Abdel-Salem, G. M., Babul-Hirji, R., Baxter, P., Berger, A., Bonafé, L., Brunstom-Hernandez, J. E., Buckard, J. A., Chitayat, D., … Crow, Y. J. (2012). Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature genetics, 44(3), 338–342. https://doi.org/10.1038/ng.1084
- Polvi, A., Linnankivi, T., Kivelä, T., Herva, R., Keating, J. P., Mäkitie, O., Pareyson, D., Vainionpää, L., Lahtinen, J., Hovatta, I., Pihko, H., & Lehesjoki, A. E. (2012). Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. American journal of human genetics, 90(3), 540–549. https://doi.org/10.1016/j.ajhg.2012.02.002
- Cela, P., Hampl, M., Shylo, N. A., Christopher, K. J., Kavkova, M., Landova, M., Zikmund, T., Weatherbee, S. D., Kaiser, J., & Buchtova, M. (2018). Ciliopathy Protein Tmem107 Plays Multiple Roles in Craniofacial Development. Journal of dental research, 97(1), 108–117. https://doi.org/10.1177/0022034517732538
- Lambacher, N. J., Bruel, A. L., van Dam, T. J., Szymańska, K., Slaats, G. G., Kuhns, S., McManus, G. J., Kennedy, J. E., Gaff, K., Wu, K. M., van der Lee, R., Burglen, L., Doummar, D., Rivière, J. B., Faivre, L., Attié-Bitach, T., Saunier, S., Curd, A., Peckham, M., Giles, R. H., … Blacque, O. E. (2016). TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nature cell biology, 18(1), 122–131. https://doi.org/10.1038/ncb3273
- Parisi M, Glass I. Joubert Syndrome. 2003 Jul 9 [Updated 2017 Jun 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325/
- Turnpenny PD, Sloman M, Dunwoodie S. Spondylocostal Dysostosis, Autosomal Recessive. 2009 Aug 25 [Updated 2023 Aug 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK8828/
- Longoni M, Pober BR, High FA. Congenital Diaphragmatic Hernia Overview. 2006 Feb 1 [Updated 2020 Nov 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1359/
- Oji, V., Preil, M. L., Kleinow, B., Wehr, G., Fischer, J., Hennies, H. C., Hausser, I., Breitkreutz, D., Aufenvenne, K., Stieler, K., Tantcheva-Poór, I., Weidinger, S., Emmert, S., Hamm, H., Perusquia-Ortiz, A. M., Zaraeva, I., Diem, A., Giehl, K., Fölster-Holst, R., Kiekbusch, K., … Traupe, H. (2017). S1 guidelines for the diagnosis and treatment of ichthyoses – update. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG, 15(10), 1053–1065. https://doi.org/10.1111/ddg.13340
- Egawa, G., & Kabashima, K. (2018). Barrier dysfunction in the skin allergy. Allergology International, 67(1), 3–11. https://doi.org/10.1016/j.alit.2017.10.002
- Richard G. Autosomal Recessive Congenital Ichthyosis. 2001 Jan 10 [Updated 2023 Apr 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1420/
- Yang, P., Pardon, L. P., Ho, A. C., Lauer, A. K., Yoon, D., Boye, S. E., Boye, S. L., Roman, A. J., Wu, V., Garafalo, A. V., Sumaroka, A., Swider, M., Viarbitskaya, I., Aleman, T. S., Pennesi, M. E., Kay, C. N., Fujita, K. P., & Cideciyan, A. V. (2024). Safety and efficacy of ATSN-101 in patients with Leber congenital amaurosis caused by biallelic mutations in GUCY2D: A phase 1/2, multicentre, open-label, unilateral dose escalation study. The Lancet, 404(10456), 962–970. https://doi.org/10.1016/S0140-6736(24)01447-8
- Tsang, S. H., & Sharma, T. (2018). Leber congenital amaurosis. In S. H. Tsang & T. Sharma (Eds.), Atlas of Inherited Retinal Diseases (Vol. 1085, pp. 131–137). Springer International Publishing. https://doi.org/10.1007/978-3-319-95046-4_26
- Kumaran N, Pennesi ME, Yang P, et al. Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy Overview. 2018 Oct 4 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531510/
- Lin, M.-H., Chou, P.-C., Lee, I.-C., Yang, S.-F., Yu, H.-S., & Yu, S. (2023). Inherited reticulate pigmentary disorders. Genes, 14(6), 1300. https://doi.org/10.3390/genes14061300
- Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. 2009 Nov 12 [Updated 2023 Jan 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22301/
- Turner JT, Brzezinski J, Dome JS. Wilms Tumor Predisposition. 2003 Dec 19 [Updated 2022 Mar 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1294/
- Schneider K, Zelley K, Nichols KE, et al. Li-Fraumeni Syndrome. 1999 Jan 19 [Updated 2024 Sep 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1311/
- Van Tol, W., Ashikov, A., Korsch, E., Abu Bakar, N., Willemsen, M. A., Thiel, C., & Lefeber, D. J. (2019). A mutation in mannose‐phosphate‐dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Reports, 50(1), 31–39. https://doi.org/10.1002/jmd2.12060
- Sparks SE, Krasnewich DM. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. 2005 Aug 15 [Updated 2017 Jan 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1332/
- Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
- Abicht A, Müller JS, Lochmüller H. Congenital Myasthenic Syndromes Overview. 2003 May 9 [Updated 2021 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1168/
- Degtyareva, A. V., Nikitina, I. V., Orlovskaya, I. V., Zakharova, E. Y., Baidakova, G. V., Ionov, O. V., … & Levadnaya, A. V. (2016). Very long-chain acyl-coenzyme A dehydrogenase deficiency. Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics), 59(4), 41-47.
- Mendez-Figueroa, H., Shchelochkov, O. A., Shaibani, A., Aagaard-Tillery, K., & Shinawi, M. S. (2010). Clinical and biochemical improvement of very long-chain acyl-CoA dehydrogenase deficiency in pregnancy. Journal of perinatology : official journal of the California Perinatal Association, 30(8), 558–562. https://doi.org/10.1038/jp.2009.198
- Leslie ND, Saenz-Ayala S. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2009 May 28 [Updated 2023 Jul 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6816/
- Rodríguez-Palmero, A., Boerrigter, M. M., Gómez-Andrés, D., Aldinger, K. A., Marcos-Alcalde, Í., Popp, B., Everman, D. B., Lovgren, A. K., Arpin, S., Bahrambeigi, V., Beunders, G., Bisgaard, A. M., Bjerregaard, V. A., Bruel, A. L., Challman, T. D., Cogné, B., Coubes, C., de Man, S. A., Denommé-Pichon, A. S., Dye, T. J., … Tümer, Z. (2021). DLG4-related synaptopathy: a new rare brain disorder. Genetics in medicine : official journal of the American College of Medical Genetics, 23(5), 888–899. https://doi.org/10.1038/s41436-020-01075-9
- Tümer Z, Dye TJ, Prada C, et al. DLG4-Related Synaptopathy. 2023 Jun 22. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK592682/
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
- Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021).
- Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2021).
- Cheng, J et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science (2023).







