
文章摘要
猫鸣综合征是一种罕见的遗传病,其特征是在婴儿期发出类似猫叫的高音哭声。这种疾病是由于第5号染色体短臂(5p)部分缺失引起的,可能导致多种身体、发育及行为问题。通过早期诊断与综合干预,可显著改善患者的发育和生活质量。
这个基因座上高度可能涉及疾病的基因

| 序号 | 基因名称 | 相关疾病名称(中文) | 相关疾病名称(英文) |
|---|---|---|---|
| 1 | DLG4 | DLG4相关突触病 | DLG4-Related Synaptopathy |
| 2 | ACADVL | 极长链酰基辅酶A脱氢酶缺乏症 | Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency |
| 3 | CHRNB1 | 先天性肌无力综合征 | Congenital Myasthenic Syndrome |
| 4 | MPDU1 | N型糖链合成及多条代谢通路的先天性疾病 | Congenital Disorders of N-Linked Glycosylation and Multiple Pathways |
| 5 | TP53 | Wilms肿瘤易感综合征、Li-Fraumeni综合征 | Wilms Tumor Predisposition; Li-Faumeni Syndrome |
| 6 | WRAP53 | 先天性角化不良症及相关端粒生物学疾病 | Dyskeratosis Congenita and Related Telomere Biology Disorders |
| 7 | GUCY2D | Leber先天性黑朦、早发型严重视网膜营养不良 | Leber Congenital Amaurosis; Early-Onset Severe Retinal Dystrophy |
| 8 | ALOX12B | 常染色体隐性先天性鱼鳞病 | Autosomal Recessive Congenital Ichthyosis |
| 9 | ALOXE3 | 常染色体隐性先天性鱼鳞病 | Autosomal Recessive Congenital Ichthyosis |
| 10 | HES7 | 先天性膈疝、脊肋骨发育不良 | Congenital Diaphragmatic Hernia; Spondylocostal Dysostosis |
| 11 | TMEM107 | Meckel综合征13型、Joubert综合征 | Meckel syndrome 13・Joubert Syndrome |
| 12 | CTC1 | 先天性角化不良症及相关端粒生物学疾病 | Dyskeratosis Congenita and Related Telomere Biology Disorders |
| 13 | RANGRF | Brugada综合征 | Brugada Syndrome |
| 14 | RPL26 | Diamond-Blackfan贫血 | Diamond-Blackfan Anemia |
| 15 | NTN1 | 先天性镜像动作 | Congenital Mirror Movements |
[1_DLG] DLG4 相关突触病(DLG4-Related Synaptopathy)

DLG4相关突触病是一种由DLG4基因突变引起的罕见遗传疾病。该基因编码一种重要的蛋白质——PSD-95(突触后致密蛋白95),它在大脑中调节兴奋性突触功能。该疾病的主要特征包括发育迟缓、智力障碍(多为轻度至中度)、以及自闭症谱系障碍。此外,大约一半的患者会出现癫痫,大约40%的患者会出现运动功能或语言能力的退行。
其他神经学症状包括肌张力低下(低肌张力)、重复动作、共济失调(运动协调障碍)、肌张力障碍、震颤以及偏头痛。睡眠障碍也很常见,常表现为入睡困难或睡眠维持困难。此外,还会出现斜视、远视、眼球震颤以及大脑皮质性盲等眼部异常。有时癫痫发作、晕车或疲劳可引起呕吐。身体特征上,关节过度柔韧(约37%)以及脊柱侧弯(约20%)相对常见。

目前尚无根治DLG4相关突触病的方法,治疗的重点是症状管理与生活质量改善。根据患者的个体需求,对发育迟缓、智力障碍、癫痫、偏头痛、共济失调、肌张力障碍、视力障碍等症状进行针对性干预。对于失眠,行为疗法有时有效,严重睡眠障碍时可考虑药物治疗。
疾病管理需要定期复查和监测。在就诊过程中,需要评估是否出现新的症状,例如癫痫发作、运动功能变化、肌张力变化、偏头痛等。同时还需评估发育进展、行动能力以及日常生活技能。在儿童成长过程中,焦虑、注意缺陷多动障碍(ADHD)、攻击行为和自伤行为等问题也可能出现,需要进行适当干预。眼科检查建议每年一次或按需进行;若出现发育退行或脑电图异常,则应考虑进行24小时脑电图监测(EEG)。
DLG4相关突触病通常是由于DLG4基因的新发(de novo)致病突变引起的常染色体显性遗传病。少数情况下,可能由携带嵌合型或杂合型突变的父母遗传,但家族内复发风险通常较低。通过基因检测确认致病突变,可以为未来妊娠提供产前诊断或植入前遗传学诊断的选择。
该疾病的预后(寿命)尚不明确,但已有47岁生存的病例报告,提示患者有生存至成年期的可能。过去曾认为该疾病有马凡样体型或特征性面容,但最新研究表明这些并非常见特征。大多数病例的发病机制与DLG4基因突变导致的蛋白功能缺失有关。临床表现和病情严重程度在不同个体间差异较大,主要影响大脑的发育与功能。
[2_ACADVL] 极长链酰基辅酶A脱氢酶缺乏症(Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency, VLCAD缺乏症)

极长链酰基辅酶A脱氢酶缺乏症(VLCAD缺乏症)是一种罕见的遗传性疾病,其特征是在线粒体内无法正常分解长链脂肪酸。这一分解过程对于在禁食或机体能量需求增加的情况下生成能量至关重要。该疾病由ACADVL基因的突变引起,该基因编码负责脂肪酸β氧化初始步骤的酶。当该酶缺陷存在时,有害的脂肪酸代谢物会在体内积聚,可能损伤心脏、肝脏、骨骼肌等多个器官。
根据严重程度和发病年龄,VLCAD缺乏症可以分为三种主要类型:
- 早发型(心脏和多器官功能衰竭型)
这是最为严重的类型,通常在出生后的数个月内发病。常见症状包括肥厚型或扩张型心肌病、心包积液、心律失常、肌张力低下(低肌张力)、肝肿大(肝脏肿大),以及无酮症低血糖(低酮性低血糖)。 - 肝型(低酮性低血糖型)
通常在幼儿期发病,主要表现为低血糖和肝肿大,但不伴有心肌病。 - 晚发型(肌肉型)
在青少年或成人期发病,主要表现为运动后出现的肌肉疼痛、痉挛、运动耐力下降,以及由运动或禁食诱发的横纹肌溶解症(肌肉组织分解)。此类型的患者在发作期间很少表现出低血糖。
随着新生儿筛查(NBS)的普及,VLCAD缺乏症的早期诊断率显著提高。许多患者在诊断时无症状,早期管理的启动可以有效预防严重并发症并改善预后。据估计,VLCAD缺乏症的发病率约为每30,000至100,000个新生儿中有1例。
治疗与管理
VLCAD缺乏症的治疗核心是预防代谢危象并管理症状。
- 日常护理
日常护理包括采用低长链脂肪、高中链脂肪酸(MCT)的特殊饮食方案。通过这种饮食调整,可以绕过缺陷的酶途径,为身体提供替代能源。同时,为防止空腹引发症状,尤其在婴幼儿阶段,推荐进行频繁进食,并在夜间提供含复杂碳水化合物的加餐。
有时还会补充左旋肉碱、欧米伽脂肪酸以及维生素A、D、E等营养补充剂。对于容易因过度运动诱发症状的患者,运动量的科学管理尤为关键。 - 急诊处理
在急性期,需通过静脉补充葡萄糖和液体维持血糖水平稳定,并防止横纹肌溶解症。如果已发生横纹肌溶解症,应立即进行充分的液体补充及尿液碱化,以保护肾功能。若患者需要接受手术,应进行术前的周密代谢管理,以降低并发症风险。

监测与随访
有效的管理依赖于定期监测。主要包括:
- 生长和发育进程的评估
- 营养状态的定期检查
- 血液检测以监测代谢标志物
- 心脏功能评估,包括超声心动图
- 对肌肉健康状态的定期监测
遗传学与风险
VLCAD缺乏症遵循常染色体隐性遗传模式。这意味着如果父母双方都是ACADVL基因突变的携带者,每次怀孕时孩子患病的概率为25%。通过基因检测可以识别携带者,并为未来妊娠提供产前诊断或植入前遗传学诊断的选择。
预后
如果进行正确的诊断与个体化管理,即使是重症型婴儿,也有望获得良好的预后。随着筛查技术、饮食管理以及医疗干预的进步,越来越多的患者可以享有较健康的生活。然而,该疾病的有效控制依赖于终生监测和个体化的治疗方案。
[3_CHRNB1] 先天性肌无力综合征(Congenital Myasthenic Syndrome, CMS)

先天性肌无力综合征(CMS)是一种罕见的遗传性疾病,会影响神经与肌肉之间的信号传递,从而引起肌无力。这种肌无力通常会在活动过程中加重。症状多在出生后或幼儿早期(主要在2岁以内)出现,但也有少数病例在成人期发病。CMS主要影响骨骼肌,不会累及心肌和平滑肌。智力、协调性、感觉和腱反射通常保持正常。然而,某些CMS亚型除了肌无力,还会表现出更复杂的症状。
CMS的严重程度和进展在不同亚型之间差异很大。有的人仅表现为轻度的运动不耐受,而有的人则会出现显著的肌无力,甚至因发热、感染或应激而突然出现呼吸衰竭。在新生儿期,常见症状包括间歇性呼吸暂停、发绀、喂养困难、吸吮力弱、哭声微弱以及全身性肌无力。一些病例中,由于胎儿期活动不足,还会出现关节挛缩(称为先天性关节挛缩症),或表现为细长的面容、狭窄的下颌、高拱腭等特征性面部外貌。在幼儿期以后,常见症状包括运动发育迟缓、上睑下垂(眼皮下垂)、眼外肌无力、面部及咽喉肌群无力,以及吞咽或发声困难。
一些患者会出现称为“肢带型肌无力”的特定表现,主要影响腰部或肩部周围的肌肉,导致步态不稳或上下楼梯困难。这些症状在压力、感染或发热期间可能突然加重,因此需要谨慎管理。
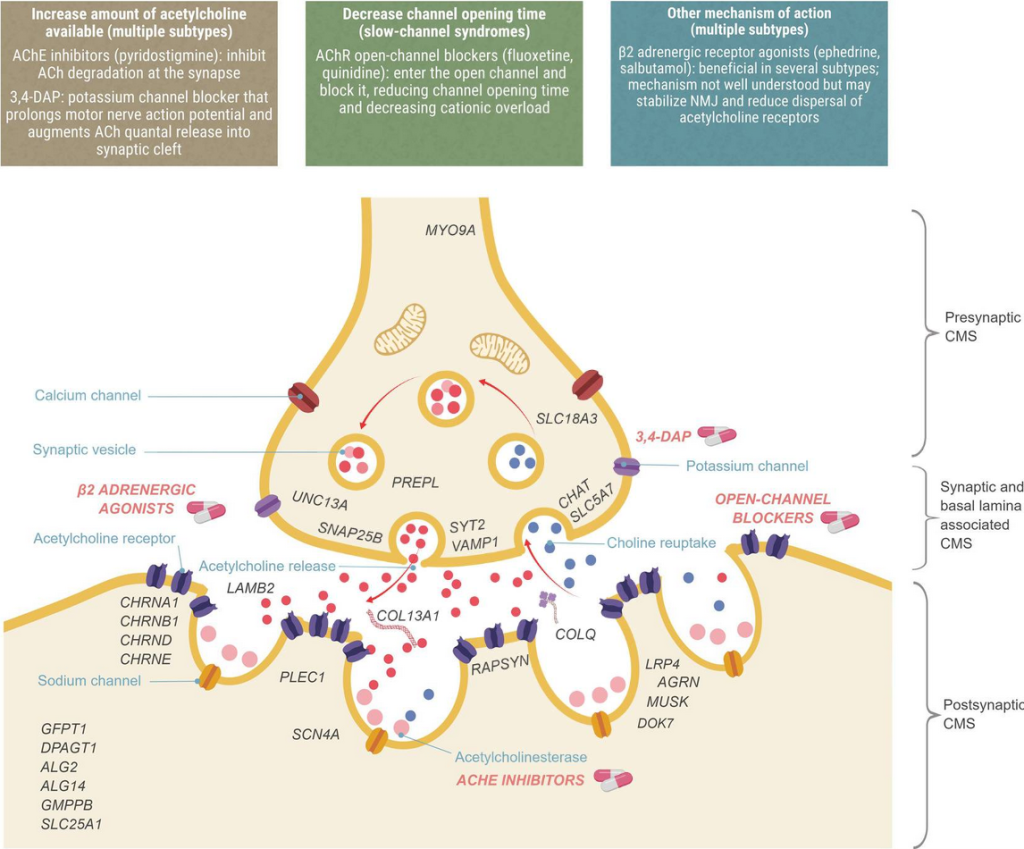
Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). [Figure 1. Localization of CMS types and therapeutic strategies.] Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
治疗与管理
CMS的治疗高度个体化,需根据患者具体情况及亚型决定。基因检测在制定治疗方案中至关重要,因为不同亚型对药物反应差异明显。常用的药物包括吡啶斯的明(乙酰胆碱酯酶抑制剂),用于改善神经与肌肉之间的信号传递,但并非所有亚型都对其有效,部分情况下甚至会加重症状。其他选择包括3,4-二氨基吡啶(3,4-DAP,可增加神经递质释放)以及麻黄碱或沙丁胺醇(在部分特定亚型中有效)。药物副作用需要严密监测,并根据个体反应调整剂量。对于CHRNB1相关的CMS,氟西汀、奎尼丁和吡啶斯的明被推荐作为一线用药,沙丁胺醇或麻黄碱可作为辅助治疗。在开始任何药物治疗前,务必咨询专业医生。
除了药物治疗,CMS的管理还应包括多学科支持,以提高生活质量。物理治疗、作业治疗及语言治疗均有帮助;在病情较重的患者中,可能需要轮椅、矫形支具或呼吸辅助装置。若存在明显的进食困难,可能需要胃造口。对于婴幼儿,建议父母配备呼吸暂停监测器,并接受心肺复苏术(CPR)培训,以应对突发的呼吸危机。
随访与监测
肌力、呼吸功能以及整体健康状况需要定期随访。通常儿童每6个月复查一次,成年人每年复查一次。孕期患者应与专科医生密切合作,监测呼吸及心脏功能。
其他注意事项
由于CMS并非由免疫反应引起,免疫抑制治疗无效。然而,大多数CMS亚型对某些药物治疗有一定反应,但效果因亚型不同而显著差异。某些药物在特定亚型中有效,但在其他亚型中可能引发不良反应。因此,精确诊断并制定个体化治疗方案至关重要。在科学管理和规律监测下,CMS患者能够有效控制症状,显著改善生活质量。
[4_MPDU1] N型糖链附加与多通路的先天性疾病(Congenital Disorders of N-Linked Glycosylation and Multiple Pathways)

N型糖链型先天性糖基化异常症以及多通路异常症(统称为先天性糖基化障碍,Congenital Disorders of Glycosylation, CDG)是一组由糖基化修饰过程异常导致的罕见遗传性疾病。糖基化是指糖分子被附着到蛋白质或脂质上的过程,对细胞功能极其重要,其异常会影响多器官,造成多样化的临床症状。
CDG通常在婴儿期起病,典型表现包括生长不良、发育迟缓、肌张力低下、肝脏异常,以及神经系统问题如癫痫。还常见眼部异常、皮肤问题如鱼鳞病和免疫功能障碍。亚型之一的MPDU1-CDG(又称CDG-If)由参与糖分子跨膜转运的MPDU1基因异常引起,该过程是糖基化中的关键步骤。
MPDU1-CDG是CDG中极为罕见的一型。CDG属于多系统性遗传病群,由糖蛋白合成过程的缺陷引发。这些缺陷导致血清糖蛋白糖基化不足,从而损害对胚胎发生、细胞分化及细胞功能维持至关重要的N型糖蛋白作用,进而引发全身性影响,产生多彩而复杂的临床表现。
MPDU1-CDG由MPDU1基因变异导致。该基因在细胞内负责特定糖分子的转运,是糖基化关键步骤之一。该功能受损会使正确糖基化的蛋白和脂质无法生成,出现发育迟缓、肌张力低下、癫痫、干燥或发红的皮肤改变(如鱼鳞样改变),以及面部特征异常等。有些患儿还会出现呼吸暂停,因此呼吸管理十分重要。
MPDU1-CDG的严重程度和进展差异很大,取决于个体与变异类型。常见全身性问题包括精神运动发育落后、神经学障碍、凝血异常、免疫缺陷和皮肤异常等。糖基化异常可对神经、骨骼、免疫等多个系统产生复杂影响

治疗以对症支持为主。特别是婴幼儿期的营养管理至关重要,若存在喂养困难或生长障碍,可需要特殊配方奶、营养补充或管饲。若因口腔运动障碍导致呕吐,可通过增稠饮食或制酸剂加以处理。结合言语、物理与作业治疗有助于改善发育迟缓与运动功能。
由于眼部异常常见,应早期就诊眼科以保护视力。肾与甲状腺功能亦可能受累,建议定期肾脏超声与尿检以早期发现肾病综合征或蛋白尿。术前需评估潜在的凝血异常,必要时给予血制品或血浆输注。
急性期如卒中样发作或严重感染时,应迅速给予静脉输液与血糖管理等支持。对婴幼儿,呼吸暂停管理尤其关键,建议监护人接受呼吸急救的训练。

确诊依赖基因检测,并有助于与其他CDG亚型鉴别。早期诊断可实现更恰当的干预,改善预后与生活质量。由于疾病罕见,长期数据有限,但已有病例在综合照护下存活至青春期。
MPDU1-CDG的治疗需多学科合作,包括神经、消化、眼科、血液科与免疫科等共同管理,以应对疾病带来的多重挑战,提升整体疗效。
[5_TP53] Wilms肿瘤易感综合征、Li-Fraumeni综合征(Wilms Tumor Predisposition; Li-Fraumeni Syndrome)

Li-Fraumeni综合征(LFS)是一种显著提高青少年期发癌风险的罕见遗传性疾病,呈常染色体显性遗传,因此患者将变异传递给子代的概率为50%。LFS主要与TP53基因的生殖系变异相关。TP53对细胞分裂调控与抑癌至关重要。
LFS的特征常见于家族内多发肿瘤。经典诊断标准包括:45岁前的肉瘤患者(指示病例),其一级亲属中有人在45岁前罹患任一癌症,且同一或另一位近亲中有45岁前肿瘤或任何年龄的肉瘤病例。未完全符合标准但高度可疑者称为Li-Fraumeni样综合征(LFL)。与LFS高度相关的肿瘤包括乳腺癌、软组织与骨肉瘤、脑肿瘤(如星形细胞瘤)与肾上腺皮质癌,这些约占TP53变异者肿瘤的80%。此外还见白血病、结直肠癌、胃癌、Wilms肿瘤、恶性叶状肿瘤、脉络丛乳头状瘤等。

Wilms肿瘤是主要发生于儿童的肾癌,为LFS相关肿瘤之一。常以看似健康儿童的腹部包块起病,可能伴腹痛、发热、贫血、血尿与高血压。约5%至10%为双侧或多发性,遗传素因如LFS会提高发生率。诊断需组织学分析,分良好与退行型两类。退行型常伴体细胞TP53变异,预后较差。治疗包括手术、化疗,必要时放疗。尽管总体生存率超过90%,退行型或远处转移者预后较差。

LFS的肿瘤风险随年龄而变。儿童期常见肾上腺皮质癌、横纹肌肉瘤与脑肿瘤,青少年与年轻成人期以乳腺癌、肉瘤与脑肿瘤为主,成年后期胰腺癌与前列腺癌风险上升。终生发癌风险女性约90%,男性约70%,约半数在40岁前发病。受治疗影响与遗传素因,二次肿瘤风险亦升高。
管理包括预防、监测与治疗。女性可考虑预防性乳腺切除以降低风险。监测建议频繁的体检、全身MRI及结合家族史与年龄的器官特异性筛查。放疗会提高二次癌风险,应尽量避免。遗传咨询对受累家族十分重要,TP53检测可识别携带者并实现早期干预。
妊娠期女性需谨慎监测肿瘤迹象。虽对TP53变异胎儿无特定产前监测建议,但建议出生后即开始筛查。罕见并发症包括妊娠性绒毛癌。
LFS凸显个体化医疗与积极管理的重要性,以改善患者与家族的预后。
[6_WRAP53] 先天性发育不良及相关端粒生物学疾病(Dyskeratosis Congenita and Related Telomere Biology Disorders)

Dyskeratosis Congenita(DC)及相关端粒生物学障碍(TBD)是一组由于端粒维持异常而致的极罕见遗传病。端粒保护染色体末端,对基因稳定性与细胞寿命至关重要。多种基因变异,尤其是WRAP53,会破坏此过程并引发多样临床表现与并发症。
WRAP53位于染色体17p13,编码端粒酶Cajal小体蛋白1(TCAB1)。该蛋白在端粒合成、DNA修复与抑癌蛋白p53的调节中发挥要役。p53通路对于细胞周期与DNA损伤应答不可或缺。WRAP53变异导致端粒维持缺陷,可引起常染色体隐性型的DC亚型(DKCB3),显示端粒维持异常对多个器官系统的广泛影响。
DC/TBD临床表现高度异质。DC的经典三联征为胸颈部网状色素沉着、指甲发育不良、口腔白斑,但并非人人具备,个体差异很大。可见进行性骨髓衰竭、骨髓增生异常综合征、急性白血病、鳞癌(尤以头颈部与肛生殖部位)、肺纤维化与肝病等。其他还包括流泪、眼睑内外翻、牛牙样根形态(taurodontism)、胃肠毛细血管扩张、股骨或肩部缺血性坏死。早发白发、骨质疏松、再障亦常见,程度因人而异。多在5至10岁起病,常因骨髓衰竭、感染、肺并发症或恶性肿瘤而早逝。
DKCB3为DC的一型,遗传方式为常染色体隐性。整体上DC可呈X连锁隐性、常染色体显性或隐性遗传。疾病多见于男性,但各族群均有报道。根本原因是端粒维持异常,抑制上皮与造血细胞增殖,导致细胞衰老。色素异常被认为与老化黑色素细胞中黑色素合成增加相关。多数患者运动发育与神经功能正常,但部分可见发育迟缓。

DC/TBD治疗复杂且需个体化。造血干细胞移植是骨髓衰竭或白血病的唯一根治治疗,但长期毒性风险高。无合适供者时,可考虑雄激素治疗以改善骨髓功能。其他并发症如恶性肿瘤与肺纤维化需对症处理,重度肺纤维化时可需肺移植。肿瘤治疗增加长期血细胞减少与器官毒性风险。
随访管理至关重要,建议每年血细胞计数与骨髓检查、肿瘤筛查、肺功能与肝功能评估。接受雄激素治疗者需特别监测肝与内分泌。为减少口腔白斑与继发感染风险,建议每6个月口腔检查并保持良好口腔卫生。避免吸烟、过度日晒与部分药物可降低肿瘤与并发症风险。
DC/TBD极罕见,一般人群发病率不明。至2022年3月,全球记录病例约800至1,000例,凸显其需要专业化照护与进一步研究。
[7_GUCY2D] Leber先天性黑朦、早发型重度视网膜营养不良(Leber Congenital Amaurosis; Early-Onset Severe Retinal Dystrophy)

Leber先天性黑朦1型(LCA1)由位于17p13.1的GUCY2D基因变异引起。该基因编码视细胞杆锥体中重要的网膜鸟苷酸环化酶1(RETGC-1),负责生成cGMP,参与光信号转导中光响应恢复的关键过程,亦可能参与蛋白向外节的运输。RETGC-1失常会致视细胞功能障碍与严重变性。
LCA1通常在出生或幼年早期起病,属于“Leber先天性黑朦(LCA)”与“早发重度视网膜营养不良(EOSRD)”谱系中尤为严重的一端。其特征包括自出生即重度视力低下或失明、眼球震颤、瞳孔对光反应减弱或消失,以及揉、按、戳眼的“眼数字征”。还可见畏光、重度远视、圆锥角膜和随时间进展的视网膜色素改变。电生理(ERG)多几乎无反应。

婴幼儿期眼底可能外观正常,随龄出现色素性视网膜病变、血管狭窄、黄斑萎缩与视神经苍白。亦可伴视神经乳头玻璃样小体与晶状体混浊。尽管视功能严重受损,OCT常显示视网膜结构相对保存,这提示恢复RETGC-1功能的治疗可能改善视力。
LCA总体发生率约1/80,000,GUCY2D变异占全部LCA的6%至21%。遗传方式多为常染色体隐性。LCA1的视力通常介于无光感与约0.05之间,自然恢复可能性低。
管理需多学科协作,自早期即介入发育支持与低视力康复,辅以教育与康复资源及视力辅助装置。
近年基因治疗取得进展,例如向视细胞递送GUCY2D正常拷贝以恢复RETGC-1功能。诸如ATSN-101等新疗法为LCA1患者带来希望,有望维持或改善视力。
[8_ALOX12B,9_ALOXE3] 常染色体隐性先天性鱼鳞病(Autosomal Recessive Congenital Ichthyosis)


常染色体隐性先天性鱼鳞病(ARCI)是一组由皮肤屏障形成相关基因变异引起的罕见遗传性皮肤病,其中ALOX12B与ALOXE3尤为重要。变异会阻碍形成与维持皮肤屏障所需酶的功能。ARCI特征为先天或婴幼儿早期出现的广泛性鳞屑性脱屑与红斑。亚型包括板层型鱼鳞病(LI)与先天性鱼鳞样红皮病(CIE)等。
ALOX12B编码12-脂氧合酶,参与角质细胞脂质包膜形成,对ω-羟基神经酰胺修饰至关重要,从而维持皮肤保湿与屏障防护。ALOX12B变异导致ARCI2,出生时常见薄而有光泽的“羊皮膜”,随后脱落并发展为LI或CIE的鳞屑与红斑。多数ALOX12B变异者表现为细小鳞屑与红斑的轻中度CIE。
ALOXE3编码过氧化物异构酶,位于ALOX12B下游,同属脂质修饰通路,生成辅助屏障形成的脂质衍生物。ALOXE3功能缺失致ARCI3,临床上兼具LI与CIE特征。约三分之一ALOXE3变异者出生时有羊皮膜,此后多表现为轻中度CIE。
ARCI新生儿因屏障受损,面临脱水、体温调节障碍、感染与呼吸并发症高风险。羊皮膜会随着时间剥落,转变为全身性鳞屑与红斑。重症的哈雷昆鱼鳞病形成厚板样皮与深裂隙,可危及生命,需重症新生儿护理与系统性维甲酸治疗。

管理聚焦缓解屏障损伤的影响,包括高湿度环境保湿、卫生管理预防感染、疼痛与代谢管理。早期使用保湿剂可强化屏障并降低并发症与特应性皮炎风险。

轻型ARCI随时间可改善,但多遗留干燥、轻度鳞屑与角化过度。重型如哈雷昆型可能长期存在炎症、耐热差、关节挛缩与眼部异常。成人期皮肤癌风险可能增加,需终生监测与护理。
诊断需基因检测、制定个体化治疗方案,并推进针对根本遗传或生化缺陷的治疗研究。新生儿护理进步与早期干预改善了预后。
[10_HES7] 先天性膈疝、脊肋骨发育不良(Congenital Diaphragmatic Hernia; Spondylocostal Dysostosis)

脊肋骨发育异常症4型(SCDO4)与先天性膈疝(CDH)与HES7基因变异相关。HES7编码转录因子HES-7,调控DNA转录,参与中胚层多种结构的发育,并作为转录抑制因子作用于含N盒与E盒的启动子。其功能受损可致严重发育异常。

SCDO4因胚胎期分节异常致椎肋畸形,表现为躯干相对矮短、短颈、轻度不进展性脊柱侧弯、椎体融合、半椎体、肋骨后方融合、肋骨数目减少或形态异常。新生儿可因胸廓小而呼吸受限,重症需重症监护,但约至2岁肺生长改善,部分可较正常发育。重症仍可并发慢性呼衰、肺高压与心脏问题。男性SCDO患者腹股沟疝风险高,需及时处理。
诊断依赖X线显示分节异常与肋骨畸形,并通过包括HES7在内的相关基因致病变异确诊。治疗包括重度侧弯的外科干预、急慢性呼衰的呼吸支持,以及对生长、脊柱弯曲、肺功能与神经问题的定期监测。因SCDO由HES7变异致常染色体隐性遗传,遗传咨询、携带者检测与产前诊断可行。超声可在妊娠约13周检测脊柱异常。

CDH为HES7相关SCDO少数病例的合并症,表现为横膈形成或肌化不全,横膈组织缺损或不足,或横膈变薄并异常抬高。多在出生时即显现,发生率约为每1万人3至3.6例。HES7相关SCDO中CDH不足10%。新生儿期需立刻给予适当呼吸支持以应对致命性呼衰。
总体显示,HES7变异强调早期胚胎发育与转录调控对椎、肋与横膈形成的关键作用。诊断、管理与咨询缺一不可。
[11_TMEM107] Meckel综合征13・Joubert综合征(Meckel syndrome 13・Joubert Syndrome)

Meckel综合征13由TMEM107变异引起,极罕见且严重,常染色体隐性遗传。主要特征为囊性肾病、脑发育畸形(尤以枕部脑膨出)与因板状形成异常致肝纤维化。还可见多指、小脑蚓部发育不全。Meckel综合征致死性极高,多在胎内或生后不久死亡。
TMEM107编码跨膜蛋白107,参与纤毛形成与功能。纤毛调控发育关键信号通路,如SHH、WNT、PDGF,影响神经、骨骼、颜面与颅骨发育。TMEM107变异致纤毛数目减少,出现异常细长扭曲等缺陷,引发Meckel综合征的重度发育障碍。
Meckel综合征属于“纤毛病”谱系,其中包括Joubert综合征(JS)。JS较Meckel轻,部分可存活至儿童或成人期,但在脑畸形、多指、肝肾异常等方面高度相似。同一家族内可有个体表现为Meckel,另有个体为JS,提示两者为同一谱系不同严重度。通常功能完全缺失的变异致Meckel,较轻变异致JS。
JS特征为影像学“磨牙征”、发育迟缓、肌张力低下与婴儿期呼吸异常。尚可见视力障碍、语言迟缓与广泛的智力差异。TMEM107相关JS的亚型“口面指综合征(JS-OFD)”出现舌肿瘤、腭裂、口腔黏膜皱襞等,使与Meckel的界限更为模糊。

Meckel综合征治疗以对症为主,因致死率高。已知风险家族应优先考虑无创产前检测(NIPT)等早期诊断。虽无治愈方法,但对相关纤毛病如JS的特定并发症干预可获益,包括对呼吸异常、运动问题与发育迟缓的治疗,以及对腭裂、多指、肝肾异常的外科或内科处理。存活者需多学科照护。
总体预后严峻,反映纤毛功能障碍对早期发育的深远影响。
[12_CTC1] 先天性发育不良及相关端粒生物学疾病(Dyskeratosis Congenita and Related Telomere Biology Disorders)

2023年有一例38岁已婚女性携带CTC1基因区域两处变异,被诊断为Coats plus综合征的报道。此罕见遗传病伴随复杂医疗挑战,但在恰当治疗、照护与支持下,受累儿童仍可带着希望生活。

Coats plus综合征,正式名称为“脑视网膜微血管病伴钙化与囊肿1型(CRMCC1)”,由CTC1基因变异导致的罕见隐性遗传病。其发病涉及分别自父母继承的两个异常等位基因。CTC1是CST复合体的组成成分,负责稳定端粒并辅助DNA准确复制。CTC1变异使复合体功能失常,端粒缩短、染色体易损,进而引发疾病表现。
该病累及神经系统及全身。神经方面有脑内钙化、白质损伤与脑囊肿,导致痉挛、共济失调、肌张力障碍、癫痫与认知下降。眼部方面常见类似Coats病的视网膜血管扩张与渗出。
全身表现包括骨质疏松导致骨脆,胃肠与肝脏血管异常引起消化道出血与门脉高压。还可见毛发、皮肤、指甲异常,以及贫血与血小板减少。
由于与其他端粒相关病如“异常角化症”共享特征,需尽早筛查骨髓功能低下、肿瘤、肺与肝病等并发症。与Labrune综合征的鉴别要点包括是否有系统性表现。
研究显示,异常蛋白导致CST复合体功能障碍,推进端粒缩短与染色体异常,推动疾病进展与症状加重。
总之,Coats plus综合征是一种复杂疾病,涵盖眼病、脑钙化与损伤、骨脆、消化道问题等多方面。其病理基础是CTC1变异致端粒保护受损,强调CST复合体的重要性。
[13_RANGRF] Brugada综合征(Brugada Syndrome)

Brugada综合征是导致心脏电活动异常的遗传性疾病,可引发生命危险的心律失常与猝死。最常相关的基因为SCN5A,编码维持心脏节律的钠通道Nav1.5。RANGRF基因同样重要,编码MOG1蛋白,作为辅助因子帮助Nav1.5正确定位于心肌细胞膜并发挥功能。RANGRF变异会妨碍Nav1.5膜定位,降低钠通道功能,导致心律异常与猝死风险上升。
Brugada综合征通常在成人期起病,亦可见于婴儿至老年。猝死平均年龄约40岁。特征性心电图表现为V1至V3导联ST段异常与心室性心律失常风险增高。临床症状包括晕厥、婴儿猝死综合征,以及主要见于东南亚的夜间猝死综合征。男性患病率约为女性的8倍,儿童期极罕见。
诊断基于特征性心电图所见,可自然显现或药物诱发。结合个人与家族史,若发现SCN5A或其他相关基因致病变异则确诊。
治疗以预防猝死为目标。有晕厥或心脏骤停史者,植入式心律转复除颤器(ICD)是最有效治疗。高风险患者可辅以奎尼丁以减少心律失常与ICD放电。一些病例中,右室流出道心外膜消融是有前景的选择。对有家族史或已知致病变异的高风险人群,建议定期心电图监测。

预防方面,避免高热、特定药物与可诱发心律失常的物质十分重要。
Brugada综合征多为常染色体显性遗传,即受累父母的子女有50%概率遗传。外显率不完全与表现差异显著,携带变异者未必发病。少数为新生突变。建议受累家族进行遗传咨询,若已明确致病变异,可进行产前或植入前遗传学检测。
全球患病率估计约1/2,000,但地区与族群差异显著。亚洲与中东较高,西班牙裔与白人较少。多数患者无症状,约30%有晕厥,8%至12%可发生心脏骤停。早期识别与干预至关重要。
[14_RPL26] Diamond-Blackfan贫血(Diamond-Blackfan Anemia)

Diamond-Blackfan贫血11(DBA11)由RPL26基因变异引起。RPL26编码核糖体结构成分,参与蛋白合成。RPL26功能缺失会导致核糖体功能障碍,从而引发DBA11。DBA是婴儿期内出现的再生障碍性贫血。
主要特征为中至重度贫血,常见红细胞体积偏大。骨髓中红系前体细胞显著减少,而白细胞与血小板通常正常。30%至40%患者伴生长障碍或先天畸形,包括颌面异常(如Pierre Robin序列、腭裂)、拇指异常、泌尿生殖系异常。重症可因胎儿期贫血出现胎儿水肿。DBA还增加某些肿瘤风险,如急性髓系白血病、骨髓增生异常综合征与骨肉瘤。

诊断依据生后一年内出现的大细胞性贫血、网织红细胞减少、骨髓红系前体缺乏,并排除其他原因。分子诊断通过检测与DBA相关的致病变异。多数为常染色体显性遗传,也有GATA1或TSR2变异导致的X连锁遗传。
治疗以改善贫血与预防并发症为中心。12个月以上儿童对糖皮质激素常有疗效,但长期使用风险包括感染、成长抑制与骨密度下降。对激素无效或副作用显著者,需反复红细胞输注或造血干细胞移植(HSCT)。HSCT是治愈DBA贫血的唯一方法。骨、眼与内分泌问题需多学科处理。肿瘤治疗时需谨慎使用化疗以避免血细胞进一步下降。
输血相关铁过载等次生并发症使用铁螯合治疗。需定期监测铁负荷与激素副作用。随访包含血液与骨髓评估以及肿瘤筛查。分子遗传学检测可识别高危家族并实现早期干预。

DBA主要为常染色体显性遗传,40%至45%病例自父母遗传,其余为新生突变。X连锁情形下,携带变异的男性将变异传给全部女儿而非儿子;女性携带者传递概率为50%,儿子受遗传则发病,女儿多为携带者。
妊娠期应密切监测母体血红蛋白,必要时使用低剂量阿司匹林以降低并发症风险。建议进行遗传咨询,并可选择产前或植入前诊断。
DBA表明核糖体功能与全身发育密切相关,核糖体缺陷可导致血液异常与多系统先天畸形。
[15_NTN1] 先天性镜像运动(Congenital Mirror Movements)

镜像运动4型(MRMV4)由NTN1基因变异引起。NTN1编码网蛋白-1,参与神经轴突向导与细胞存活。网蛋白-1引导中枢交联轴突与外周运动轴突生长,通过与DCC与UNC5家族受体结合,调控轴突的“吸引”与“排斥”。例如与UNC5C结合可使受体与微管分离,增强微管动态并促进排斥。网蛋白-1还通过抑制凋亡促进神经细胞存活,与肿瘤发生发展亦相关。

网蛋白-1功能丧失可致MRMV4,呈常染色体显性遗传。其特征为一侧随意运动在对侧出现镜像的非自主动作。幼儿期可有一过性镜像运动,但超过7岁仍存在则属异常。MRMV4多累及上肢,尤以手指与手部动作最明显。步行通常不受影响,但足趾肌肉可轻度受累。
先天性镜像运动通常自幼出现,贯穿成年,不进展亦不改善。强度个体差异明显,通常弱于刻意动作。部分人因重复手工或持续动作出现上肢疼痛,影响精细作业与单手任务。脑MRI通常正常,但若由DCC变异所致,可见胼胝体部分或完全缺如。迄今尚无因NTN1变异而报告胼胝体异常的病例。

治疗以适应为主,因其非进展性。对儿童可在教育上给予延长考试时间、减少书写任务等照顾。青少年与成人宜选择不需复杂双手协调或长时间手工的职业。为减少社会偏见并发挥个体潜能,心理支持亦重要。
该病呈常染色体显性遗传,但外显率可能较低,因此即使遗传了致病变异也未必发病。未发病父母亦可能将变异传给子女。若已明确致病变异,可选择产前或植入前诊断。
患病率极低,估计低于1/1,000,000,但轻症漏诊多,实际患病率可能更高。主要致病基因为NTN1、DCC或RAD51,其中以DCC相关最常见,外显率约42%。
未来展望
随着DNA测序技术的进步,无创产前检测(NIPT)作为替代传统FISH与羊水穿刺等侵入性方法的高可靠且安全的筛查手段已逐渐普及。羊水穿刺需用针穿刺子宫获取羊水或绒毛细胞,而NIPT通过分析母体血液中的胎儿游离DNA进行评估,不增加母体与胎儿风险,因而成为越来越多孕妇的选择。
即便检出遗传异常,许多情况下,孩子依然有机会成长并拥有充实的人生。随着研究进展,这种可能性在不断扩大。期待NIPT带来的早期发现成为获得安心与为家庭未来做好准备的助力,而非新的不安来源。

参考文献
- Méneret A, Trouillard O, Dunoyer M, et al. Congenital Mirror Movements. 2015 Mar 12 [Updated 2020 Sep 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK279760/
- Janov, A. J., Leong, T., Nathan, D. G., & Guinan, E. C. (1996). Diamond-Blackfan anemia. Natural history and sequelae of treatment. Medicine, 75(2), 77–78. https://doi.org/10.1097/00005792-199603000-00004
- Willig, T.-N., Niemeyer, C. M., Leblanc, T., Tiemann, C., Robert, A., Budde, J., Lambiliotte, A., Kohne, E., Souillet, G., Eber, S., Stephan, J.-L., Girot, R., Bordigoni, P., Cornu, G., Blanche, S., Guillard, J. M., & Mohandas, N. (1999). Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 diamond-blackfan anemia patients. Pediatric Research, 46(5), 553–553. https://doi.org/10.1203/00006450-199911000-00011
- Sieff C. Diamond-Blackfan Anemia. 2009 Jun 25 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7047/
- Marfatia, K. A., Harreman, M. T., Fanara, P., Vertino, P. M., & Corbett, A. H. (2001). Identification and characterization of the human MOG1 gene. Gene, 266(1-2), 45–56. https://doi.org/10.1016/s0378-1119(01)00364-x
- Brugada R, Campuzano O, Sarquella-Brugada G, et al. Brugada Syndrome. 2005 Mar 31 [Updated 2022 Aug 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1517/
- Campuzano, O., Berne, P., Selga, E., Allegue, C., Iglesias, A., Brugada, J., & Brugada, R. (2014). Brugada syndrome and p.E61X_RANGRF. Cardiology journal, 21(2), 121–127. https://doi.org/10.5603/CJ.a2013.0125
- Kayarian, F. B., Cohen, S. M., Cohen, M. L., & Sammartino, D. E. (2023). Coats plus syndrome presenting in an adult. Journal of VitreoRetinal Diseases, 7(6), 562–564. https://doi.org/10.1177/24741264231171465
- Anderson, B. H., Kasher, P. R., Mayer, J., Szynkiewicz, M., Jenkinson, E. M., Bhaskar, S. S., Urquhart, J. E., Daly, S. B., Dickerson, J. E., O’Sullivan, J., Leibundgut, E. O., Muter, J., Abdel-Salem, G. M., Babul-Hirji, R., Baxter, P., Berger, A., Bonafé, L., Brunstom-Hernandez, J. E., Buckard, J. A., Chitayat, D., … Crow, Y. J. (2012). Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nature genetics, 44(3), 338–342. https://doi.org/10.1038/ng.1084
- Polvi, A., Linnankivi, T., Kivelä, T., Herva, R., Keating, J. P., Mäkitie, O., Pareyson, D., Vainionpää, L., Lahtinen, J., Hovatta, I., Pihko, H., & Lehesjoki, A. E. (2012). Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. American journal of human genetics, 90(3), 540–549. https://doi.org/10.1016/j.ajhg.2012.02.002
- Cela, P., Hampl, M., Shylo, N. A., Christopher, K. J., Kavkova, M., Landova, M., Zikmund, T., Weatherbee, S. D., Kaiser, J., & Buchtova, M. (2018). Ciliopathy Protein Tmem107 Plays Multiple Roles in Craniofacial Development. Journal of dental research, 97(1), 108–117. https://doi.org/10.1177/0022034517732538
- Lambacher, N. J., Bruel, A. L., van Dam, T. J., Szymańska, K., Slaats, G. G., Kuhns, S., McManus, G. J., Kennedy, J. E., Gaff, K., Wu, K. M., van der Lee, R., Burglen, L., Doummar, D., Rivière, J. B., Faivre, L., Attié-Bitach, T., Saunier, S., Curd, A., Peckham, M., Giles, R. H., … Blacque, O. E. (2016). TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nature cell biology, 18(1), 122–131. https://doi.org/10.1038/ncb3273
- Parisi M, Glass I. Joubert Syndrome. 2003 Jul 9 [Updated 2017 Jun 29]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1325/
- Turnpenny PD, Sloman M, Dunwoodie S. Spondylocostal Dysostosis, Autosomal Recessive. 2009 Aug 25 [Updated 2023 Aug 17]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK8828/
- Longoni M, Pober BR, High FA. Congenital Diaphragmatic Hernia Overview. 2006 Feb 1 [Updated 2020 Nov 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1359/
- Oji, V., Preil, M. L., Kleinow, B., Wehr, G., Fischer, J., Hennies, H. C., Hausser, I., Breitkreutz, D., Aufenvenne, K., Stieler, K., Tantcheva-Poór, I., Weidinger, S., Emmert, S., Hamm, H., Perusquia-Ortiz, A. M., Zaraeva, I., Diem, A., Giehl, K., Fölster-Holst, R., Kiekbusch, K., … Traupe, H. (2017). S1 guidelines for the diagnosis and treatment of ichthyoses – update. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG, 15(10), 1053–1065. https://doi.org/10.1111/ddg.13340
- Egawa, G., & Kabashima, K. (2018). Barrier dysfunction in the skin allergy. Allergology International, 67(1), 3–11. https://doi.org/10.1016/j.alit.2017.10.002
- Richard G. Autosomal Recessive Congenital Ichthyosis. 2001 Jan 10 [Updated 2023 Apr 20]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1420/
- Yang, P., Pardon, L. P., Ho, A. C., Lauer, A. K., Yoon, D., Boye, S. E., Boye, S. L., Roman, A. J., Wu, V., Garafalo, A. V., Sumaroka, A., Swider, M., Viarbitskaya, I., Aleman, T. S., Pennesi, M. E., Kay, C. N., Fujita, K. P., & Cideciyan, A. V. (2024). Safety and efficacy of ATSN-101 in patients with Leber congenital amaurosis caused by biallelic mutations in GUCY2D: A phase 1/2, multicentre, open-label, unilateral dose escalation study. The Lancet, 404(10456), 962–970. https://doi.org/10.1016/S0140-6736(24)01447-8
- Tsang, S. H., & Sharma, T. (2018). Leber congenital amaurosis. In S. H. Tsang & T. Sharma (Eds.), Atlas of Inherited Retinal Diseases (Vol. 1085, pp. 131–137). Springer International Publishing. https://doi.org/10.1007/978-3-319-95046-4_26
- Kumaran N, Pennesi ME, Yang P, et al. Leber Congenital Amaurosis / Early-Onset Severe Retinal Dystrophy Overview. 2018 Oct 4 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK531510/
- Lin, M.-H., Chou, P.-C., Lee, I.-C., Yang, S.-F., Yu, H.-S., & Yu, S. (2023). Inherited reticulate pigmentary disorders. Genes, 14(6), 1300. https://doi.org/10.3390/genes14061300
- Savage SA, Niewisch MR. Dyskeratosis Congenita and Related Telomere Biology Disorders. 2009 Nov 12 [Updated 2023 Jan 19]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK22301/
- Turner JT, Brzezinski J, Dome JS. Wilms Tumor Predisposition. 2003 Dec 19 [Updated 2022 Mar 24]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1294/
- Schneider K, Zelley K, Nichols KE, et al. Li-Fraumeni Syndrome. 1999 Jan 19 [Updated 2024 Sep 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1311/
- Van Tol, W., Ashikov, A., Korsch, E., Abu Bakar, N., Willemsen, M. A., Thiel, C., & Lefeber, D. J. (2019). A mutation in mannose‐phosphate‐dolichol utilization defect 1 reveals clinical symptoms of congenital disorders of glycosylation type I and dystroglycanopathy. JIMD Reports, 50(1), 31–39. https://doi.org/10.1002/jmd2.12060
- Sparks SE, Krasnewich DM. Congenital Disorders of N-Linked Glycosylation and Multiple Pathway Overview. 2005 Aug 15 [Updated 2017 Jan 12]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1332/
- Thompson, R., Bonne, G., Missier, P., & Lochmüller, H. (2019). Targeted therapies for congenital myasthenic syndromes: systematic review and steps towards a treatabolome. Emerging topics in life sciences, 3(1), 19–37. https://doi.org/10.1042/ETLS20180100
- Abicht A, Müller JS, Lochmüller H. Congenital Myasthenic Syndromes Overview. 2003 May 9 [Updated 2021 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1168/
- Degtyareva, A. V., Nikitina, I. V., Orlovskaya, I. V., Zakharova, E. Y., Baidakova, G. V., Ionov, O. V., … & Levadnaya, A. V. (2016). Very long-chain acyl-coenzyme A dehydrogenase deficiency. Rossiyskiy Vestnik Perinatologii i Pediatrii (Russian Bulletin of Perinatology and Pediatrics), 59(4), 41-47.
- Mendez-Figueroa, H., Shchelochkov, O. A., Shaibani, A., Aagaard-Tillery, K., & Shinawi, M. S. (2010). Clinical and biochemical improvement of very long-chain acyl-CoA dehydrogenase deficiency in pregnancy. Journal of perinatology : official journal of the California Perinatal Association, 30(8), 558–562. https://doi.org/10.1038/jp.2009.198
- Leslie ND, Saenz-Ayala S. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2009 May 28 [Updated 2023 Jul 13]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK6816/
- Rodríguez-Palmero, A., Boerrigter, M. M., Gómez-Andrés, D., Aldinger, K. A., Marcos-Alcalde, Í., Popp, B., Everman, D. B., Lovgren, A. K., Arpin, S., Bahrambeigi, V., Beunders, G., Bisgaard, A. M., Bjerregaard, V. A., Bruel, A. L., Challman, T. D., Cogné, B., Coubes, C., de Man, S. A., Denommé-Pichon, A. S., Dye, T. J., … Tümer, Z. (2021). DLG4-related synaptopathy: a new rare brain disorder. Genetics in medicine : official journal of the American College of Medical Genetics, 23(5), 888–899. https://doi.org/10.1038/s41436-020-01075-9
- Tümer Z, Dye TJ, Prada C, et al. DLG4-Related Synaptopathy. 2023 Jun 22. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK592682/
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
- Jumper, J et al. Highly accurate protein structure prediction with AlphaFold. Nature (2021).
- Varadi, M et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research (2021).
- Cheng, J et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science (2023).
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。







