

Overview
Currarino Syndrome (CS) is a rare genetic disorder characterized by the “Currarino triad,” which includes three main features: sacral anomalies, sacral anterior meningocele, and anorectal malformations. The severity of symptoms varies significantly between individuals, and although the prognosis is generally favorable, early diagnosis and proper management are crucial. Prenatal diagnostics, including non-invasive prenatal testing (NIPT), are discussed for early detection.
Synonyms of Currarino Syndrome
- Currarino Triad
- Currarino Idiopathic Osteoarthropathy
- Currarino Disease
- Cranio-Osteoarthropathy
- Reginato-Schiapachasse Syndrome
- Curras
Disease Overview

Currarino syndrome is a rare hereditary disease affecting 1 to 9 individuals per 100,000 population, with a slightly higher prevalence in females (1.39 females for every male). It results from a mutation in the MNX1 gene located on chromosome 7. This mutation impairs the proper development of certain structures during the early stages of pregnancy.
Key Features (Currarino Triad):
Sacral Abnormalities: These occur in the sacrum (the triangular bone at the base of the spine). The sacrum may not develop properly, and parts of it may be missing. The first sacral vertebra (the uppermost sacral bone) may have an abnormal shape.
Sacral Anterior Meningocele: This is a tumor or cyst-like mass in front of the sacrum, which can vary in size and type. It may cause local pressure and can result in neurological issues or problems with bowel and bladder control.
Anorectal Malformations: These involve abnormal development of the anus and rectum, leading to issues with defecation.
Additional symptoms can include chronic constipation, neonatal bowel obstruction, infections around the anus, kidney and urinary tract problems, reproductive system abnormalities in females, and spinal tethering (spinal cord being abnormally pulled by surrounding tissue) or meningomyelocele (spinal cord bulging at the tip).

Inheritance and Genetic Basis
Currarino syndrome follows an autosomal dominant inheritance pattern, meaning if one parent carries a mutation in the MNX1 gene, there is a 50% chance that the gene will be passed on to the offspring. However, the manifestation and severity of the condition can vary greatly, even within the same family, due to other genetic factors.
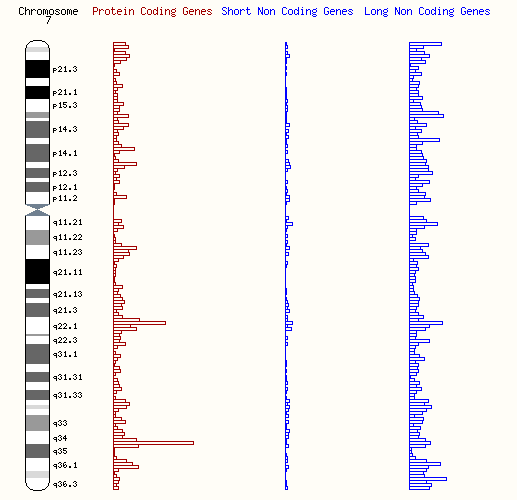
The genetic mutation is located on chromosome 7 (7q36.3), and mutations in the MNX1 gene are the primary cause. MNX1 is involved in early developmental stages, particularly the development of the spinal cord and other critical structures.
Diagnosis
Diagnosis is based on clinical features, supported by imaging techniques like X-rays, CT scans, and MRIs, which can reveal sacral anomalies and anterior meningocele. Genetic testing for the MNX1 mutation can also confirm the diagnosis.

Management and Treatment
Treatment depends on the severity and type of abnormalities present. Surgical repair of anorectal malformations is often needed. Conservative management may include medications to relieve constipation or follow-up for monitoring. For sacral anterior meningocele, observation is often recommended unless intervention is required. If the spinal cord is involved, neurosurgical interventions may be necessary.
Long-term Prognosis
Many individuals with Currarino syndrome have a good long-term prognosis, though some experience ongoing issues such as constipation, urinary incontinence, and neurological symptoms. Malignant transformation of sacral tumors is rare, but it can occur in a small percentage of cases. Regular follow-up is necessary to monitor and manage these potential issues.
Prenatal Diagnosis
Prenatal diagnosis of Currarino syndrome can be challenging, but it is possible, especially when there is a known family history of the condition. Non-invasive prenatal testing (NIPT) has emerged as a reliable screening method, providing accurate results without risk to the mother or fetus. Invasive techniques such as amniocentesis (for DNA analysis) or FISH testing can also be used, but these carry some risks to both the mother and child.
NIPT has gained popularity among pregnant women due to its safety and reliability in detecting genetic conditions. For those at risk, carrier screening before pregnancy can help make informed decisions about family planning and appropriate care for any affected children.

Genetic Counseling and Screening
For families with a known history of Currarino syndrome, genetic counseling is recommended. Testing family members for the MNX1 mutation can provide valuable information for reproductive decision-making.
In conclusion, Currarino syndrome is a complex genetic disorder with a range of symptoms. Early diagnosis, whether through clinical examination or prenatal testing, along with proper management, can significantly improve outcomes. Non-invasive prenatal testing offers a safe and reliable option for families seeking early diagnosis and peace of mind during pregnancy.

reference
- Ferreira, C., Santos, A. P., & Fonseca, J. (2022). Currarino syndrome – a pre and post natal diagnosis correlation: Case report and literature review. The Journal of Maternal-Fetal & Neonatal Medicine, 35(25), 5224–5226. https://doi.org/10.1080/14767058.2021.1876021
- Yoshida, A., Maoate, K., Blakelock, R., Robertson, S., & Beasley, S. (2010). Long-term functional outcomes in children with Currarino syndrome. Pediatric Surgery International, 26(7), 677–681. https://doi.org/10.1007/s00383-010-2615-4
- Ross, A. J., Ruiz-Perez, V., Wang, Y., Hagan, D.-M., Scherer, S., Lynch, S. A., Lindsay, S., Custard, E., Belloni, E., Wilson, D. I., Wadey, R., Goodman, F., Orstavik, K. H., Monclair, T., Robson, S., Reardon, W., Burn, J., Scambler, P., & Strachan., T. (1998). A homeobox gene, HLXB9, is the major locus for dominantly inherited sacral agenesis. Nature Genetics, 20(4), 358–361. https://doi.org/10.1038/3828
- Holland, P. W., Booth, H. A. F., & Bruford, E. A. (2007). Classification and nomenclature of all human homeobox genes. BMC Biology, 5(1), 47. https://doi.org/10.1186/1741-7007-5-47
- Dworschak, G. C., Reutter, H. M., & Ludwig, M. (2021). Currarino syndrome: A comprehensive genetic review of a rare congenital disorder. Orphanet Journal of Rare Diseases, 16(1), 167. https://doi.org/10.1186/s13023-021-01799-0
- Orphanet. (Last updated April 2023). Currarino syndrome. Reviewed by Dr John COLEMAN & Prof. Sally Ann LYNCH. Retrieved from https://www.orpha.net/en/disease/detail/1552
- Online Mendelian Inheritance in Man. (Last updated September 2007 by Cassandra L. Kniffin). Retreived from https://omim.org/entry/176450
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。







