

L. Martínez-Jacobo a C. Córdova-Fletes a, b R. Ortiz-López a, b F. Rivas cC. Saucedo-Carrasco d A. Rojas-Martínez a, b
a 医学部生化学・分子医学科、b 分子生物学部、 基因组学和测序学、 蒙特雷的新莱昂州自治大学健康科学研究开发中心、 c 奥克西登特综合医院、哈利斯科州卫生部、瓜达拉哈拉事务局、以及 d 私人办公室、小儿科、圣路易斯波托西、墨西哥
关键词
阵列CGH ・ 候选基因 ・ Delineation del(7q) ・ 手裂足裂领域
要约
在这项研究中,我们介绍了一名女性患者,其通过G‐带和CGH‐SNP阵列测定了7q21.3q31.1的出生时有一个新的缺失。 该患者表现出精神运动迟缓、先天性严重的双侧青光眼、腭裂和心脏缺陷等特征。 微阵列检测显示,缺失的12.5Mb领域,在与手指和脚趾畸形有关的区域约88kb的下游;因此,患者的最终核型为46,XX.arr 7q21.3q31.1(96,742,140~109,246,085)×1dn。这个女孩是迄今为止记载的第4名患者,先天性青光眼以及包括7q21.3q31.1领域在内的重复缺失,证实了与这种临床特征相关的基因或者基因的存在。 根据作者的结果,继发于7q中间缺失的眼部缺陷的趋势,可能是由TAC1、HBP1、及细胞色素P450基因的小集群(亚家族3A)的同时缺失造成的。 这一结论得到了功能作用和发现部位的支持。另外,TAC1具有与青光眼相关的突变的MYOC基因的功能路径相关。 此外,鉴于女孩在临床上回忆起与7q21q32内多种缺失相关联的几种表型,通过我们的结果和观察表明,确认了7q21.3q31.1处重复缺失/表型的基因内容提供一般的概观,CUX1基因以及DLX5开始的下游的潜在的调节原件在内的DLX基因远位的座位,与手指和脚趾畸形无关。
著作权 2013 S. Karger AG, Basel
序言
先天性7q缺失是相当常见的,但也是异质性的。 这些缺失作为全体包含7q(DECIPHER数据库),通常分类为近端(q11→21)、中间(q21→31/32)以及末端(q32→qter),[Gibson等、1982、Young等、1984、Chong等、2008]。
L.M.-J.与C.C.-F.对这项研究的贡献相同。7q21q32段内的各种缺失,常常导致手指和脚趾畸形和多种临床特征,如智力障碍/发育迟缓、耳/听觉异常、低出生体重、进食困难、异常哭泣、小头症、小颚畸形、心脏和腭部缺陷、反复感染、异常的手掌皱纹以及眼睛异常[Young等、1984、Riviera等、1991、Scherer等、1994、Montgomery等、2000、Bernardini等、2008、Chong等、 缺失区域的明显重叠与表型的一致性,实际上可能代表一种可识别的缺失综合症[Fagan等 1989]。 在本文中,我们做了一个完善中间(q21→31/32)缺失相关联的眼睛的表现型的图谱并提供进一步的洞察力,7q21.3q31.1缺失以及先天性严重青光眼(不伴有手指和脚趾畸形)的报告。
患者记述
患者是一个1岁的女孩,是没有血缘关系的父母所生的第2个孩子。没有异常的家族病史。 妊娠晚期发现胎儿发育迟缓,短股骨和骨盆石灰化,由于胎儿风险增加,在妊娠35周时剖腹产。 出生体重为2,300g(<3百分值)、身高43cm(<3百分值),只提到阿普加评分为8分(总分10分)。先天性青光眼被诊断出来通过2次流出道重建(小梁切开术、纤维切除术)进行治疗。 病人的眼压仍然很高。 头部MRI,显示蛛网膜下腔水肿,骨髓增生过程的变化和双侧视神经水肿。 7个月大时,她的体重是4,300g(10~25百分值),身高57.3cm(<3百分值)。 她表现为眼睛大,巩膜呈蓝色,眼窝大,眼窝开放,眉部突出,耳廓过度折叠耳廓低,眼睑裂外翻(掉眼),眼角分离明显,鼻宽,鼻翼发育不良,鼻翼发育不全,鼻柱短,两侧单掌裂和两侧第5指尖部位(图1A)ーーー>鼻底与上唇基底和中线分离,嘴唇薄,上唇二重弓形(线),软腭裂,小颌症,两侧猴纹,两侧第5指尖部侧方弯曲,(图1A) 另外,还有卵圆孔开放,动脉管开放和三尖瓣反流。在1岁时,还没有颈定。 对听觉刺激的反应很弱,但她对光的刺激有反应。 牙齿没有发育,哭声微弱,体重、身高和前后头径周长分别为5,500g(不满3百分值)、67.5cm(<3百分值、42cm(<3百分值)。 眼科检查发现眼球突出,眼压升高,蓝色巩膜,角膜浑浊伴随角膜水肿为特征的两侧原发性先天性青光眼。 包括虹膜在内的葡萄膜结构显然是正常的,但不知道它们是否被植入虹膜并从虹膜血管获得血流。 病人目前正在从频繁的呼吸道感染中恢复。
材料和方法
染色体分析
对患者及其父母的初步细胞遗传学分析,是在72小时淋巴细胞培养得到的GTG带的中期染色体上进行的。 在informed consent的情况下,从患者和家长那里进一步提取的血液样本,被用于分子研究。
阵列CGH
首先,患者及其父母的基因组DNA,通过使用Qiagen Gentra® Puregene Blood core Kit从3ml的末梢血中得到。中密度微阵列分析,使用Agilent SurePrint G3 Hmn CGH+SNP 4x180K微阵列试剂盒(中央值25kb的间隔有约120,000个的CGH探针和60,000个的SNP探针)进行实施。 简单地说,患者和他的父母和性别匹配的对照组的基因组DNA(约1mg),用37℃2小时,AluI以及RsaI限制性酶(Promega, Madison, Wisc, USA)来消化,消化的生成物,用Sure tag DNA标记试剂盒(Agilent Technologies),对Cy3-dU 标记的生成物,按照Agilent的协议进行纯化、杂交和洗涤。 各玻片在Nimblegen MS 200扫描仪(Roche)上扫描,所得图像用图像转换软件进行转换,并用Feature Extraction软件(Agilent科技)进行成像。 结果,使用Agilent Cytogenomics软件v.2.5的ADM-2畸变算法,默认分析方法-CGH+SNP v2进行分析。
结果
患者的G带核型为46,XX,del(7)(q22q22)dn,父母核型正常。 相应的微阵列检测显示,包括500的标记和229的基因(miRNA以及虚拟蛋白质)在内,缺失12.5-Mb领域(基因组位置96,742,140-109,246,085),ACN9的近位C7orf66的远位(GRCh37/hg19);因此,对于7q21.3q31.1的部分单倍体的缺失,缺失指形质的关键领域[Scherer他、1994]重合,作为这样的临床特征的主要备选的DLX5以及DLX6基因不含在内。 没有观察到其他明显的基因组改变。 父母的微阵列结果是正常的。 患者最终的核型为46,XX.arr 7q21.3q31.1(96,742,140~109,246,085)×1dn。
考察
在这项研究中,我们报告了全球第4例,包括或重复的于q21.3q31.1领域与出生时伴随新的缺失有关的先天性青光眼患者。
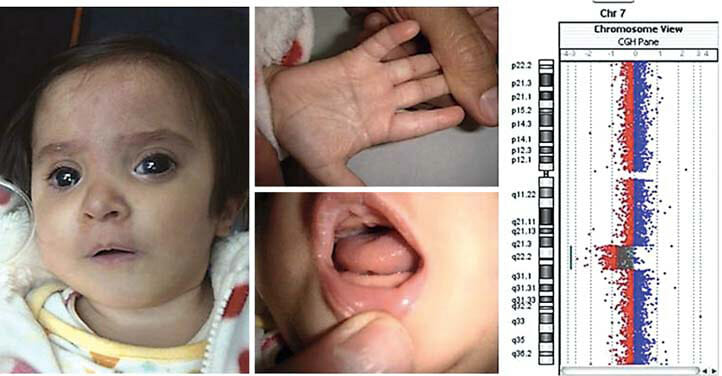

图1
A 颅面、手、口 患者的特征。
B 患者特征 log 2 ratio显示(图和线)表示。软件v.2.5进行可视化。
C 图(非比例 眼球有异常情况和无异常情况,以及7q21.3q31.1重复的7q缺失的携带者。显示7q21.3q31.1重复的7q缺失的携带者。这些染色体畸变中的大多数没有经过分子阐述,因此我们从最初分配的断点中近似地找出最初分配的断点(换句话说,我们把断点放在带的近端或远端)。
当前案例」下面的案例,DECIPHER的案例中,与这一次的缺失大体上相重合。与当前的删除重合。垂直蓝线,表示潜在的最小眼球异常区域。表示潜在的眼球异常的最小区域。最初的蓝线(左)和虚线表示,与我们的案例重复的区域。灰色线条用灰色划定了一些重复的区域。* 在这个病例中,既没有眼球异常,也没有拇指外翻的报告。是7q21q31)。* * 不正确的断点 (没有拇指外翻)。? = 不明眼球异常 没有对胎儿进行剖检,所以对眼球的影响是未知的。
D UCSC浏览截图 DECIPHER与ISCA中报告的所有不平衡案例, DECIPHER与ISCA报告的7q21.3q31.1领域重复, 7q21.3q31.1领域重复,包含DECIPHER与ISCA中报告的所有不平衡案例的浏览截图(红=缺失。蓝是增幅)。) ISCA的2个缺失 (ID是nssv578163与nssv582318。) 看似完全跨越7q21.3q31.1领域,但实际上对应的是7号染色体的单倍体。
表1. 从7q21q32中报告的的缺失病例明确临床特征,与7q21.3q31.1领域明显重叠
| 临床特征 | 案例/参考文献 | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15* | 16 | 17 | 这次的案例 | |
| 一般 | ||||||||||||||||||
| 低出生体重儿 | – | + | – | – | + | – | – | + | – | + | + | + | + | + | + | – | – | + |
| ID/DD | + | + | + | + | + | + | + | + | + | + | +a | + | + | +? | +b | + | + | + |
| 生长迟缓/身材矮小 | ||||||||||||||||||
| 身高 | – | – | – | + | + | + | – | + | – | + | – | + | – | + | – | – | – | + |
| 语言延迟 | – | – | – | – | + | + | + | – | – | – | – | – | – | – | – | – | ? | + |
| 性低血压 | – | + | – | + | + | – | – | – | – | + | – | – | + | – | – | – | – | + |
| 脑波异常/发作 | – | + | – | – | + | – | – | + | – | + | + | – | – | – | – | – | – | – |
| 再发性感染症 | + | + | – | – | + | – | – | – | –? | – | – | – | – | – | – | –? | – | + |
| 头部/眼窝 | ||||||||||||||||||
| 周围的大范围/显著的额头 | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + |
| 小头症 | + | + | – | – | + | – | – | – | – | – | – | + | – | + | – | – | + | – |
| 向上的倾斜 | – | + | – | – | – | – | – | – | – | – | + | – | – | – | – | – | – | + |
| 表情肌的褶皱 | – | – | – | + | – | – | – | + | – | – | – | – | – | – | – | – | – | NA |
| 隆起的眉毛 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + |
| Hypertelorism/眼角开离 | – | – | – | – | – | – | – | – | – | – | + | – | + | – | + | –? | + | apparently |
| 耳・听力 | ||||||||||||||||||
| Low set | + | + | – | + | – | – | – | – | – | – | + | – | + | + | – | – | + | + |
| 畸形 | + | + | – | + | + | + | + | – | – | – | + | + | + | – | – | – | – | + |
| 听觉障碍・听力障碍 | – | – | – | – | – | – | – | – | – | – | – | – | + | – | – | –? | – | apparently |
| 鼻 | ||||||||||||||||||
| 扁平/宽鼻梁 | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + | – | – | – |
| 球根状的鼻尖 | + | – | – | – | + | – | – | + | – | – | + | – | – | – | – | – | – | + |
| 眼球的异常 | + | + | G | – | – | + | – | + | + | + | G | + | + | G | ? | –? | – | G |
| 口 | ||||||||||||||||||
| 大 | + | + | – | + | + | – | + | + | – | + | + | – | + | – | – | – | – | – |
| 薄上唇 | – | – | – | + | – | – | – | + | – | ? | ? | – | ? | – | – | – | – | + |
| 长人中 | + | – | – | – | – | – | – | – | – | + | – | – | – | – | – | – | – | apparently |
| 腭裂・高腭・窄腭 | – | – | + | + | – | – | – | – | – | – | – | + | – | + | – | – | – | + |
| 小颌症 | + | + | – | – | – | – | – | – | – | + | – | + | + | – | – | – | – | + |
| 颈部营养障碍 | – | + | – | – | + | + | + | + | – | + | + | – | + | – | – | – | – | mildc |
| 颈部 | ||||||||||||||||||
| 短 | – | – | – | + | – | + | – | – | – | – | + | – | – | – | – | –d | – | – |
| 四肢・手 | ||||||||||||||||||
| 第5指的无指症 | – | + | – | – | – | – | mild | + | – | – | – | – | – | – | – | – | + | + |
| 分裂手指/分裂足 | – | – | – | – | – | – | – | – | – | – | – | + | + | + | – | – | – | – |
| 掌底皱纹异常 | – | + | – | – | + | – | – | + | – | – | + | – | – | – | + | – | – | + |
| 心血管 | ||||||||||||||||||
| ASD/VSD | – | + | + | – | – | – | – | – | – | – | + | – | + | + | – | – | – | – |
| PDA | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + |
| 肺动脉狭窄 | – | – | – | – | – | – | – | – | – | + | – | – | – | – | – | + | – | – |
| 其他 | – | – | – | – | – | – | – | – | – | + | + | – | – | – | + | – | – | +e |
| 消化道 | ||||||||||||||||||
| 疝气 | – | – | – | – | – | – | – | + | – | – | – | + | + | – | – | – | – | – |
| 泌尿系统 | ||||||||||||||||||
| 性器异常 | – | – | + | – | – | + | – | + | – | – | – | + | – | + | – | + | – | – |
1 Ayraud et al. [1976]; 2 Higginson et al. [1976]; 3 Dennis等 [1977]; 4 Hull et al. [1979]; 5 Klep-de Pater et al. [1979]; 6 Serup [1980]; 7 Abuelo and Padre-Mendoza [1982]; 8 Young等 [1984]; 9 Chitayat et al. [1988]; 10 Fagan et al. [1989]; 11 Franceschini et al. [1978]; 12 Tajara et al. [1989]; 13 Morey and Higgins [1990]; 14 Montgomery et al. [2000]; 15 Cheong et al. [2008]; 16 ID 253694; 17 ID 255298.
ID/DD = 智力障碍/发育迟缓。 NA = 不可用。 G = 青光眼。 ? =未知/不正确。
22周分娩的胎儿,除了以上无法进行进一步评估。 a 我们提案的(早期死亡)。 b 作者提案的(早期死亡)。 c 最初的吸附很差。 d 该患者出现手部前多指症。
在这次报告后,一个24周大的胎儿出现了与作者的病例非常相似的缺失,表现为口唇裂和口腭裂、高色素症、扁平的鼻梁、小颌畸形、低耳位、小阴茎以及隐睾[Chen等 2013]。 在此表中,超过30%的病例具有以下特征、ID/DD(~18/18)、眼畸形(12/18)、低出生体重(10/18)、耳畸形(10/18)、心脏畸形(9/18)、大口(9/18)、进食障碍(9/18)、低耳(8/18)、生长迟缓/身材矮小(8/18)、肌张力低下(6/18)、小头畸形(6/18)、小下颚 「哭声微弱」或「哭声异常」的特征,没有包括在本表中,但包括本表在内的大约5名患者。
(图1C、表1)显示。 作者的结果表明,在该区域映射的至少3与眼睛相关的基因位(TAC1、CYP3A43及HBP1)的共同缺失可以解释青光眼和其他眼部异常。
青光眼是一种临床和遗传上的异质性疾病,其特点是视网膜神经节细胞丧失和视神经萎缩[Izotti等 2011; Mookherjee et al., 2012]。 眼压升高的主要临床标准通常是由于睫状体网状结构对水液的阻力所致[Kennedy等 2012]。 角膜网状结构位于角膜底部周围,在调节水液流出方面发挥着重要作用[Izotti等 2011]。 在先天性青光眼患者中,经常观察到睫状体网状结构的改变。 先天性青光眼、角膜混浊、原发性开角型青光眼和青少年开角型青光眼是青光眼的一些亚型,与GLC1A或はMYOC(1q24.3)、CYP1B1(2p22.2)、以及CAV1/CAV2(7q31.2)[Stoilov et al.、1997;Alward et al.、1998;Thorleifsson et al.、2010;Kennedy等、2012;
CYP1B1基因与初级同源物有关
具有细胞色素P450依赖性代谢物的青光眼(OMIM 231300),可调节角膜透明度和房水分泌。 此外,CYP1B1被下调,在具有变异的MYOC基因的人类小柱网细胞中,CYP26B1被上调[Kennedy等、2012]。 这些事实表明,细胞色素P450s家族中的其他基因也可能参与其中。 事实上,位于7q22.1(CYP3A4、CYP3A5、CYP3A7、CYP3A43)的细胞色素是P450小规模的基因群。
事实上,在我们的患者中,大多数与青光眼、角膜混浊、巨大角膜、或瞳孔异常等其他眼部异常相关的7q的缺失,都涉及q22,并且可能涉及CYP基因[Young等、1984;Montgomery等、2000]。 值得注意的是,CYP3A43被发现在人类角膜上皮组织中有不同的表达[Turner等 2007]。 一般来说,亚家族3A的成员被发现在人类虹膜、睫状体和角膜中表达[Chan等、2008;Volotinen等、2011]。 另一个青光眼的候选基因似乎是TAC1,这在我们的病人中也缺失,最近被认为是青光眼损伤的潜在生理生物标志物。 TAC1基因,与MYOC基因的功能通路相关。MYOC基因,是一种由结节网络的压力条件诱导的糖蛋白。事实上,TAC1的表达在MYOC突变体细胞中也有强烈改变[Kennedy等 2012]。 TAC1基因(7q21.3)是precur-编码。
作为神经递质的激素Saul(UCSC基因组浏览),已被确定为人类纤维血管柱网状结构中的机械敏感基因[Kennedy等 2012]。
该患者的另一个基因也缺失,即HBP1,是一个参与WNT途径的,在视网膜、角膜和睫状体中(EMBL-EBI及GeneCards基础数据)表达的转录抑制器。 WNT途径与眼压调节有关[Kennedy等2012]。同时,HBP1(高迁移率组框转录因子)基因,是已知的视网膜损伤和炎症应激信号传导的内源性分子,与HMGB1(高迁移率组框1蛋白质)是同样的[Lee等、2012]。
尽管7q21q32缺失症的表型是多样的,但已观察到几个反复出现的临床特征(表1)。根据表1中获得的信息,至少有14个特征在患者中发现,其频率超过30%以上(表1)。例如,这个孩子,至少表现出11种这些特征,包括智力障碍、发育迟缓、生长迟缓以及颅面、心脏和眼球缺陷。此外,断点分析显示,当前缺失的近端断点位于DLX5基因88kb的下游。因为DLX5以及DLX6基因的单倍体功能不足是导致拇指外翻的一个已知原因[Scherer他, 1994; van Silfhout他, 2009]、在决定这种肢体缺陷时,DLX5及ACN9的下游(RP11-800O14与D7S618之间)的CUX1基因(Bernardini他[2008]建议)和潜在的调节元素(Tzschach他[2007]建议)被间接证实排除在外。
这些结果证实,7q21.3q31.1是脑、心脏、成长、眼睛的生理・发育很重要的基因的区域(表2),7q21.3q31.1重复缺失・表型的基因内容概况,并进一步证实DLX基因远端的基因座与拇外翻无关。
表2. 7q21.3q31.1的缺失基因,可能与其他类似缺失病例中经常出现的一些临床特征有关
| 关联 | 基因/蛋白质 | 参考文献 |
|---|---|---|
| 脑/ID/DD | ACHE、ATXNL7、BHLHA15、COG5、GPC2、MLL5*、NPTX2、NRCAM、PNPLA8、RELN、SRPK2、SYPL1、TAC1、 THAP5、TMEM130、VGF、NYAP1a | UCSC基因组浏览、Al-Hassnan等[2011] Rymen等[2012]、解读ID263273。 Vincent等[2008]、Uliana等[2010] |
| 颅面变化 | MLL5*、PLOD3**、RELN | Al-Hassnan等[2011]、Salo等[2008] |
| 听力损失 | GJC3、LHFPL3、MLL5*、PLOD3**、SLC26A4、SLC26A5 | UCSC基因组浏览、ID263273、Salo等[2008] Albert等[2006] |
| 眼睛异常 | CLDN15、CYP3A4、CYP3A43、CYP3A5、CYP3A7、HBP1、NRCAM、TAC1、TMEM130 | EMBL-EBI、Volotinen等[2011]、Lee等[2012]、Demyanenko等[2011]、Kennedy等[2012] |
| 心脏/心脏的缺陷 | MOSPD3、NPTX2、PNPLA8、SRPK2、THAP5 | UCSC基因组浏览、Paul等[2004] |
| 消化道变化/发育迟缓・身材矮小 | CLDN15、MUC3A、MUCB、MUC17、MOGAT3 | UCSC基因组浏览、Wada等[2013] |
这些关联是根据基因功能和表达位置、基因/表型的重复,或以前的关联进行的。 应该注意的是,一些基因可能与多种临床特征有关。 事实上,一些单基因病变与各种临床特征(例)似乎有关。 MLL5*或PLOD3**。
a ISCA微小缺失例(ID nssv578204)删除。
鸣谢
我们感谢患者的父母一直以来的合作,与Dr. Jose A. Paczka(眼科医生)的报告。 本研究得到了PROMEP(No)的部分支持。 103.5/11/4330, PAICYT (SA609-10) to C. Cordova-Fletes, PAICYT (No.) SA324-10、FOMIX(Convocatoria M0014-2007-2010、Reg) 068251) CIDICS L. Martinez-Jacoboは、CONACYT奖学金的支持。
参考文献
Abuelo DN, Padre-Mendoza T: 由于7q间质缺失的猫状哭声和智力低下(7q22导致了7q32)。 J Med Genet 19: 473-476 (1982)
Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, et al: SLC26A4基因经常与高加索人群中的前庭导水管扩大的非综合症性听力障碍有关。 Eur J Human Genet 14:773-779 (2006)
Al‐Hassnan ZN、Al‐Bakheet A、Abu‐Dheim N、Al‐Younes B、Colak D、Kaya N:通过阵列比较基因组杂交技术检测到的7q22.1~7q22.3的新型基质微缺失。 Am J Med Genet 155:3128-3131 (2011)
Alward W, Fingert J, Coote M, Johnson A, Lerner S, et al: 与1号染色体开角型青光眼基因(GLC1A)的突然变异有关 N Engl J Med 338:1022-1027 (1998)
Ayraud N, Rovinski J, Lambert JC, Galiana: 第7染色体长臂的间质性缺失。 Ann Genet 19:265-268 (1976)
Bernardini L, Palka C, Ceccarini C, Capalbo A, Bottilo I, et al: 染色体7q21.13-q22.1的复杂再编成,证实了一个直接的耳聋位点,并提出了一个新的候选基因。 Am J Med Genet A 146A:238-244 (2008)
Chen CP, Chang SJ, Chern SR, Wu PS, Chen YT, et al: 7qのde novo interstitial deletement(7q22.1→q31.1)的产前诊断和遗传基因521:311-315(2013)
Cheong MLJ, Tsai MS, Cortes R, Harrison MR: 第1期ー在唐氏综合症筛查中发现的7q染色体中间间质缺失。 胎儿诊断24:340-344(2008)
Chitayat D、McGillivray BC、Wood S、Kalousek DK、Langlois S、Applegarth DA:间质性7q缺失[46,XX,del(7)(pter→q21.1::q22→qter)]及β葡萄糖醛酸酶和囊性纤维化基因的位置 Am J Med Genet 31:655-661 (1988)
Demyanenko G, Riday T, Tran TS, Dalal J, Darnell EP, et al: NrCAM缺失导致丘脑皮质轴突对视觉皮层的地形定位错误,并破坏视力。 J Neurosci 31:1545-1558(2011)
Dennis NR, Neu RL, Bannerman RM: 多发性畸形婴儿中的部分7q单倍体。 Am J Hum Genet 29:37A (1977)
Fagan K, Gill A, Henry R, Wilkinson I, Carey B: 7q的间质性缺失和β-葡萄糖醛酸酶基因排除图谱的总结。 J Med Genet 26:619-625 (1989)
Franceschini P, Silengo MC, Davi GF, Santoro MA, Prandi G, Fabris C: 染色体7 46,XX,del(7)的长臂的间质性缺失(pterはq2200:q32 Hum Genet 44:345-348 (1978)
Gibson J, Ellis PM, Forsyth JS: 第7染色体的间质性缺失: 案例报告和文献综述。 Clin Genet 22:256-265 (1982)
Higginson G, Weaver DD, Magenis RE, Prescott GH, Haag C, Hepburn DJ: 多发性畸形的幼儿在染色体7号(7q-)的长臂的间质性缺失。 Clin Genet 10:307-312 (1976)
Hull DR, Kessler KK, Juberg RC: 7q间质性缺失,导致发育不良和特异的哭泣:与以前报道的7q1和7q2缺失的比较。 Am J Hum Genet 31:97A (1979)
Izzotti A, Longobardi M, Cartiglia C, Sacca SC: 小梁网中的线粒体损伤,仅发生在原发性开角型青光眼和假性剥脱性青光眼中。 PLoS 1 6e14567 (2011)
Kennedy KD, Anithachristy SA, Buie LK, Borras T: Cystatin A: 突变的肌青霉素可能与青光眼有一般相关性。 PloS 1 7:e36301 (2012)
Klep-de Pater JM, Bijlsma JB, Bleeker-Wagemakers EM, de France HF, de Vries-Ekkers CMAM: 有第7染色体长臂的不同缺失的2病症 J Med Genet 16:151-154 (1979)
Lee J, Hsiao C, Yang I, Chou MH, Wu CL, et al: High-mobility group box 1蛋白质是大鼠视网膜神经节细胞株RGC-5中的糖基化终产物诱导型血管内皮生长因子A Mall Vision18:838-850(2012)
Montgomery TL、Wyllie J、Oley C:与7q21.2-q31.2的间质性缺失相关的乳糜指症和青光眼。 Clin Dysmorphol 9:235-239 (2000)
Mookherjee S, Acharya M, Banerjee D, Bhattacharjeee A, Ray K: 在MYOC上调方面CYP1B1参与的分子基础及其在青光眼发病机制中的潜在意义 PLoS 7:e45077 (2012)。
Morey MA, Higgins RR: 伴随7q的间质性缺失的阿梅里亚综合症。 Am J Med Genet 35:95-99 (1990)
Pall GS, Wallis J, Axton R, Brownstein DG, Gautier P, et al: 含有MSP的新型跨膜蛋白在右心室发育中的作用。 基因组84:1051-1059(2004)
Rivera H, Sanchez-Corona J, Burgos-Fuentes VR, Melendez-Ruiz MJ: 7q22的缺失与缺失指数。 Genet Couns 2:27-31 (1991)
Rymen D、Keldermans L、Race V、Rregal L、Deconinck N, et al: COG5-CDG: 临床范围的扩大。 孤儿院J Reare Dis 7:94 (2012)
Salo AM, Cox H, Farndon P, Moss C, Grindulis H, et al: 由赖氨酰羟化酶3基因的突然变异引起的结缔组织疾病。 Am J Hum Genet 83:495-503 (2008)
Scherer S, Poorkaj P, Allen T, Kim J, Geshuri D, et al: 染色体7号上的常染色体显性夹板/裂足基因位点的精细制图,波段q21.3- 1. Am J Hum Genet 55:12-20 (1994)
Serup L:第7染色体长臂的间质性缺失。 Hum Genet 54:19-23 (1980)
Stoilov I、Akarsu N、Sarfarazi M:作为染色体2p21上的与GLC3A lo-cus相关的家族史中原发性先天性青光眼(葡萄球菌)主要原因的识别细胞色素P4501B1(CYP1B1)的3种不同截断突变的同定。 Hum Mol Gen 6:641–647 (1997)
Tajara EH, Varella-Garcia M, Gusson AC: 第7染色体的间质性长臂缺失以及直指症。 Am J Med Genet 32:192-194(1989)
Thorleifsson G, Walters GB, Hewitt AW, Masson G, Helgason A, et al: 与CAV1及CAV2很近的一般的变异型,原发性开角型青光眼 Nat Genet 42:906-909 (2010)
Turner H、Budak M、Akinci M、Wolosin JM:使用寡核苷酸芯片对人类结膜和角膜上皮基因表达的比较分析。 Ophthalmol Vis Sci 48:2050-2061 (2007)投资。
Tzschach A, Menzel C, Erdogan F, Schubert M, Hoeltzenbein M, et al: 通过tiling path array CGH的16 Mb的间质性染色体7q21缺失的特征 Am J Med Genet A 143:333-337 (2007)
Uliana V, Grosso S, Cioni M, Ariani F, Papa FT, et al: 染色体7的色带q22.2-q22.3上3.2Mb的微小缺失、过成长及延迟 Eur J Med Genet 53:168-170 (2010)
van Silfhout AT, van den Akker PC, Dijkhuizenet T, Verheij J, Olderode-Berends M, et al: 染色体7q异常(SHFM1)的手足分裂畸形: 作为因果机制的功能半 Eur J Hum Genet 17:1432-1438 (2009)
与自闭症和智力障碍有关VincentJB、Choufani S、Horike S、Stachowiak B、Li M等:A转位t(6;7)(p11-p12;q22):裂解点候选基因的定位和识别。 精神科Genet 18:101-109 (2008)
Volotinen M, Hakcola J, Pelkonen O, Vapaatalo H, Mmaenpa A J: 眼科用噻吗洛尔的代谢: 一种老药的新方面 基础临床药理学工具108:297-303 (2011)
和田M, 田村A, 高桥N, 津北S: 老鼠的Claudin2和15的缺失,导致肠道旁细胞性Na(+)流和营养物质运输的缺失引起营养不良而死亡。 消化内科:S0016-5085(2012)
Young RS, Weaver DD, Kukolich MK, Heerema NA, Palmer CG, et al: 第7染色体长臂的末端缺失以及间质缺失: 共有5个新的病例对象 Am J Med Genet 17:437-450 (1984)。
Zhang T, Xiang C, Gale D, Carreiro S, Wu E, Zhang EY: 在药物输送体和人类眼球屏障中的表达细胞色素P450 mRNA发现: 对眼部药药物动态的影响 Drag Metab Dispo36:1300-1307(2008).
KARGER
e-Mail karger@karger.com ww.karger.com/msy著作权 2013 S. Karger AG, Basel 1661-8769/13/0046-0285$38.00/0
Augusto Rogas Martinez、MD/DSc
分子生物学联合、基因组、科学研究中心ー、Saud市民团体、共同研究机构
Monterrey64460 (Mexico) Nuevo・León大学电子邮件・Allojastmus@gmail.com
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。



