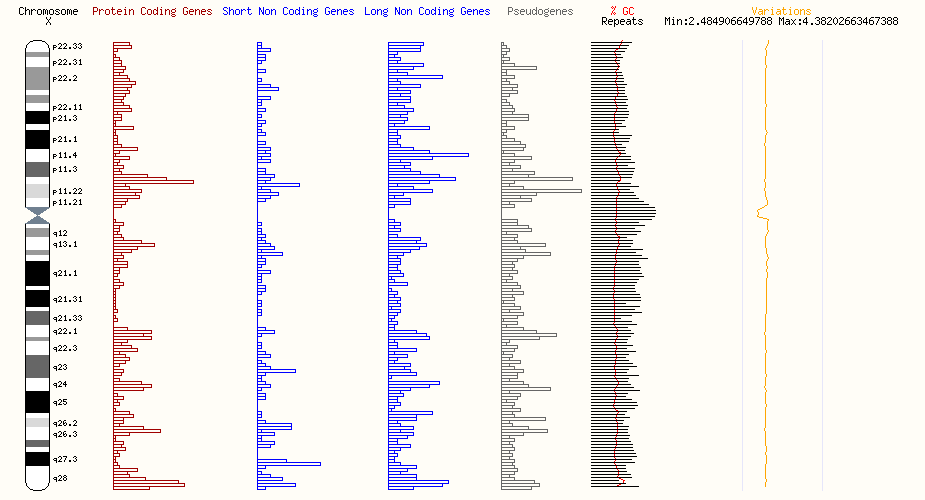
Xp11.3 欠失症候群に関する包括的な情報を提供。関連する遺伝子変異や疾患、症状、診断方法、治療アプローチについて詳しく解説します。

ダウン症の検査
気になる費用はこちら
この記事のまとめ
Xp11.3 欠失症候群は、希少疾患に分類される遺伝性疾患です。この記事では、関連する遺伝子(NDP、KDM6A、RP2など)や疾患(ノリー病、歌舞伎症候群2型、網膜色素変性症2型など)、その症状、診断、治療について詳しく解説します。
この遺伝子座にある疾患に関与する可能性が高い遺伝子

| S/N | 遺伝子名 | 関連する疾患名 | Associated disease name |
| 1 | NDP | ノリー病 | Norrie disease |
| 2 | KDM6A | 歌舞伎症候群2 | Kabuki syndrome 2 (KABUK2) |
| 3 | RP2 | 網膜色素変性症2型 | Retinitis pigmentosa 2 (RP2) |
| 4 | NDUFB11 | 多発性先天異常を伴う線状皮膚欠損 3 | Linear skin defects with multiple congenital anomalies 3 (LSDMCA3) |
| 5 | UBA1 | 脊髄性筋萎縮症X連鎖2 | Spinal muscular atrophy X-linked 2 (SMAX2) |
[1_NDP] ノリー病(Norrie disease)

ノリー病(Norrie病、ND)は、X連鎖性潜性遺伝による非常に希少な遺伝性疾患で、生後間もなく視覚障害が現れるのが特徴です。この病気は主に男性に発症し、網膜の発達に深刻な異常を引き起こします。網膜は、光や色を感じるための感覚細胞が集まる目の重要な部分ですが、ノリー病の患者では、未熟な網膜細胞が眼球後部に蓄積し、視覚機能が大きく損なわれます。この状態は「白瞳症(leukocoria)」と呼ばれ、瞳孔が光を反射して白く見える症状として現れます。さらに、病状の進行により、虹彩(目の色を構成する部分)や眼球全体が退縮することや、白内障(レンズの濁り)が進行することもあります。
ノリー病の原因は、X染色体上に位置するNDP遺伝子の変異です。この遺伝子はノリン(Norrin)というタンパク質を生成する指令を担っています。ノリンは、細胞の発達や組織の形成に重要な「Wntシグナル伝達経路」を活性化する役割を果たします。この経路は特に網膜における血管形成や感覚細胞の特化、内耳の発達、さらには体内の他の組織の形成にも関与しています。しかし、NDP遺伝子が変異すると、ノリンの機能が損なわれ、網膜や内耳の発達に必要なプロセスが正常に進まず、ノリー病の症状を引き起こします。

この病気は男性に多く見られます。これは、男性がX染色体を1本しか持たないためです。そのため、NDP遺伝子に変異がある場合、その影響が直接現れます。一方、女性はX染色体を2本持つため、片方の染色体に変異があっても、もう一方の正常な遺伝子が補助的に機能し、症状が現れにくい場合が多いです。ただし、稀に女性でも症状が見られることがあり、これは「X染色体不活性化」と呼ばれる現象により、正常な遺伝子が十分に機能しない場合に起こります。
ノリー病の主な症状には以下が挙げられます。
- 眼の症状: 網膜の異常により、網膜剥離、白内障、虹彩の萎縮、眼圧の上昇(緑内障)などが見られます。多くの患者では生後数カ月以内に視覚が完全に失われます。
- 聴覚障害: 約30%の患者が10代後半から20代にかけて感音性難聴を発症します。聴覚の低下は進行性で、補聴器や人工内耳が必要になる場合があります。
- 発達遅滞と精神的問題: 約30~50%の患者が運動スキル(例: 座る、歩く)の発達遅滞を経験し、一部の患者では精神障害(時に精神病的特徴を伴う)が見られます。
- その他の合併症: 成長障害、けいれん発作、血管系の異常(例: 下肢静脈瘤や性機能障害)が報告されています。

ノリー病の診断は、臨床症状と遺伝子検査の両方を用いて行われます。特に男性患者では、NDP遺伝子の病原性変異が確認されることで確定診断が可能です。また、保因者である女性も、稀に網膜異常や軽度の聴覚障害が認められる場合があります。
治療は主に症状の管理に焦点を当てています。眼科的な治療としては、手術やレーザー療法が検討されることがありますが、完全に視力を回復させることは難しいです。聴覚障害の進行を防ぐために補聴器や人工内耳が使用される場合があります。また、発達遅滞や精神的問題に対しては、専門家による包括的なケアが重要です。これには、早期介入プログラム、理学療法、作業療法、言語療法などが含まれます。

ノリー病は遺伝性疾患であるため、家族歴がある場合は遺伝カウンセリングが推奨されます。特に母親が保因者である場合、50%の確率で子どもに変異が遺伝する可能性があるため、出生前診断や新生児スクリーニングが重要となります。
この病気に対する根本的な治療法はまだ確立されていませんが、早期診断と適切な管理により、患者の生活の質を向上させることが可能です。現在、ノリー病に関する研究が進められており、今後より効果的な治療法が開発されることが期待されています。
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
[2_KDM6A] 歌舞伎症候群2(Kabuki syndrome 2 (KABUK2))

カブキ症候群2型(Kabuki syndrome 2, KABUK2)は、X染色体のXp11.3領域に位置するKDM6A遺伝子の変異によって引き起こされます。この遺伝子はまた、先天性横隔膜ヘルニア(Congenital Diaphragmatic Hernia)の病因にも関連しています。医学的背景がない方にも分かりやすい形で説明を試みています。
カブキ症候群2型の原因となるKDM6A遺伝子は、リジン特異的脱メチル化酵素6A(Lysine-specific demethylase 6A, KDM6A)というタンパク質をコードしています。この酵素は、ヒストンH3のリジン27(Lys-27)の脱メチル化を担い、ヒストンコードと呼ばれる遺伝子発現の制御に重要な役割を果たします。特に、三重メチル化または二重メチル化された状態のLys-27を脱メチル化する能力を持ちますが、単一メチル化された状態には影響しません。このプロセスを通じて、後部発生の調節に必要なHOX遺伝子の発現を制御します。また、Lys-27の脱メチル化は、Lys-4のメチル化と関連して行われ、PRC1複合体のリクルートとヒストンH2Aの単一ユビキチン化を調整します。さらに、この酵素は、Tボックスファミリー遺伝子の発現調節に関与するクロマチン再編成にも関わるとされています。なお、この遺伝子はX染色体の不活性化を回避する特性を持っています。

KDM6Aの機能が失われると、カブキ症候群2型が発症します。この症候群は、先天性の知的障害を特徴とする病気で、以下のような特徴がみられます。出生後の低身長、独特な顔立ち(長い眼裂、下眼瞼外側1/3の反転、広く低い鼻先、大きく目立つ耳たぶ)、口蓋裂または高い口蓋、脊椎の異常、短い第5指、胎児期指紋の持続、骨・関節のレントゲン異常、幼少期の中耳炎の反復などです。
カブキ症候群全般の臨床的特徴として、特有の顔貌(長い眼裂、弓状で広い眉、短い鼻柱と低い鼻先、大きく突出またはカップ状の耳)、軽度から中等度の知的障害、出生後の成長障害、軽度の骨格異常、胎児期指紋の持続が挙げられます。その他の特徴として、先天性心疾患、泌尿生殖器系の異常、口唇裂または口蓋裂、消化器系の異常(肛門閉鎖など)、眼瞼下垂、斜視、広く離れた歯や歯欠損などが含まれます。また、感染症や自己免疫疾患への感受性の増加、てんかん、内分泌異常(女性では早発乳房発育)、摂食困難、聴覚障害もみられることがあります。

診断は、以下の条件を基に確立されます。乳児期の筋緊張低下、発達遅延または知的障害の病歴があり、1) 特徴的な顔貌(長い眼裂と少なくとも2つ以上の特徴的顔貌)、2) KMT2DまたはKDM6Aの病的変異の存在が確認される場合です。
治療は主に症状の管理に焦点を当てます。例として、胃食道逆流症に対して食事のとろみ付けや姿勢の工夫、摂食困難が重い場合は胃瘻設置が推奨されます。認知機能の問題には心理教育的検査や特別支援教育が役立ちます。また、発作がある場合は標準的な抗てんかん治療が用いられます。心疾患がある場合は、予防的な抗生物質治療が必要となることがあります。

カブキ症候群の遺伝的要因として、KMT2D関連は常染色体顕性遺伝、KDM6A関連はX連鎖性遺伝に分類されます。前者では、新生変異による症例が多いと考えられています。後者では、母親が変異を保有する場合、男性子供が発症する可能性が高いですが、女性の場合は特徴が軽度にとどまることが一般的です。
この疾患の発生率は、日本において出生32,000人に1人と推定されています。適切な管理と支援により、患者とその家族の生活の質を向上させることが可能です。
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
[3_RP2] 網膜色素変性症2型(Retinitis pigmentosa 2 (RP2))

網膜色素変性症2型(Retinitis pigmentosa 2, RP2)は、遺伝子RP2における変異によって引き起こされる疾患で、この遺伝子は染色体Xp11.3領域に位置しています。RP2遺伝子は、XRP2と呼ばれる特殊なタンパク質をコードしています。このタンパク質は、細胞内で重要な役割を担い、ゴルジ体と線毛膜の間で物質を運搬するGTPase活性化タンパク質(GTPase-activating protein, GAP)として機能します。この輸送機能は、線毛膜上に特定のタンパク質を適切に配置するために必要です。さらに、XRP2はADPリボシル化因子様タンパク質に対するグアニンヌクレオチド解離阻害因子としても働きますが、細胞骨格の構築に関与するチューブリンの二量体化を促進する機能はありません。
RP2タンパク質は、網膜の視細胞である杆体細胞と錐体細胞で広く発現しています。視細胞内では、外節から内節、外核層(ONL)まで分布し、さらに外網状層(OPL)のシナプス末端にも見られます。加えて、内核層(INL)の双極細胞、水平細胞、アマクリン細胞といった細胞や、内網状層(IPL)、神経節細胞層(GCL)、神経線維層(NFL)にも存在することが確認されています。

RP2遺伝子が損傷を受けると、網膜色素変性症2型が発症します。この疾患は、色素性網膜症という病気の一種で、最初に杆体視細胞が機能不全に陥り、その後に錐体視細胞も損傷を受けることで進行します。初期の症状としては、夜間の視力低下を示す夜盲症や、視野の中間周辺部分の喪失が見られます。病気が進行すると、さらに遠くの視野が失われ、最終的には中心視野も消失する可能性があります。
網膜色素変性症全般の特徴として、視野の狭窄、夜盲症、そして眼底検査で観察される「骨小体様」と呼ばれる色素沈着が挙げられます。これらの症状は遺伝形式により異なり、白人集団では最も多いのが常染色体潜性(84%)で、次いで常染色体顕性(10%)、X連鎖潜性(6%)が少数派となっています。
X連鎖型網膜色素変性症(XLRP)は、脈絡網膜変性または色素性網膜症と呼ばれることもあります。この型の患者は、特に男性で、眼底に典型的な骨小体様の色素沈着や進行性の脈絡硬化を示します。これらは視力の徐々の低下を伴い、最終的に失明することも珍しくありません。女性の保因者の場合は、中年以降に視力低下や網膜異常が現れる場合がありますが、その進行や重症度は個人差があります。視力機能の予測には、光覚閾値や網膜電図(ERG)の検査が役立ちます。

RP2型の患者では、症状の現れ方に2つのタイプがあるとされています。早期発症型は重度の近視を伴い、平均して3歳頃に発症します。一方、遅発型は軽度の近視または夜盲を伴い、平均10歳頃に発症します。この違いは、遺伝子レベルではRP2遺伝子の変異が前者に、RP3遺伝子の変異が後者に関連しているとされています。
治療に関しては、進行を遅らせることを目的とした方法や、合併症の管理、心理的支援などが行われます。例えば、紫外線を防ぐ眼鏡の使用や、ビタミンAの補充療法が提案されることがあります。ただし、ビタミンA療法には肝機能の定期的な検査が必要で、特定の遺伝子変異(例: ABCA4遺伝子変異)を持つ患者には適応されません。合併症として頻繁に見られるのは白内障と黄斑浮腫で、白内障の場合は手術、黄斑浮腫の場合は薬物療法が一般的です。

心理的支援も非常に重要です。診断の時点や視力の低下が顕著になる節目で、患者が心理的な負担を軽減できるよう、専門家や患者支援団体によるサポートが提供されます。また、リハビリテーションや新たなスキル習得を支援する機関への紹介も、患者の生活の質を向上させるために有効です。
現在のところ、RP2型の根本的な治療法は確立されていませんが、遺伝子治療や細胞治療などの新しいアプローチが研究されています。患者とその家族が疾患について理解を深め、適切なケアを受けられるよう、カウンセリングを通じた支援が求められます。
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
[4_NDUFB11] 多発性先天異常を伴う線状皮膚欠損 3(Linear skin defects with multiple congenital anomalies 3 (LSDMCA3))

LSDMCA3(Linear skin defects with multiple congenital anomalies 3)は、非常に稀な遺伝性疾患で、皮膚、目、神経系、心臓などの複数の臓器や組織に異常を引き起こします。この疾患の原因は、NDUFB11という遺伝子に変異が生じることにあります。この遺伝子はX染色体上に位置し、体内でエネルギーを作り出す重要な働きをするミトコンドリアの機能に関与しています。NDUFB11がコードするタンパク質は、NADH脱水素酵素(Complex I)の一部を構成しており、この酵素はエネルギー生成の過程で電子を運ぶ役割を果たします。NDUFB11に異常があると、このエネルギー生成過程が適切に機能しなくなり、さまざまな症状が現れます。
LSDMCA3の特徴的な症状として、出生時に顔や首に見られる線状の皮膚欠損が挙げられます。これらの皮膚異常は、時間とともに自然に治癒することが多く、最終的には目立たない程度の痕跡を残すのみです。目の異常としては、涙管閉鎖、近視、斜視、眼振(目が不規則に動く症状)などが報告されています。また、心臓に関する異常も頻繁に見られ、特に心筋症(心臓の筋肉が弱くなり正常に働かなくなる病気)が多いです。神経系の異常としては、脳梁(左右の脳半球をつなぐ構造)の欠損や側脳室の拡大、けいれん発作などが挙げられます。

この疾患はX連鎖性顕性遺伝の特徴を持ちます。これは、通常男性では発症しない一方、主に女性が影響を受ける遺伝パターンを示します。稀に、軽症の母親から重症の娘が生まれるケースもあり、症状の重さには個人差が大きいことが知られています。また、家族歴がない場合でも、突然変異によって発症することがあります(de novo変異)。
臨床例として、これまでに2人の無関係な女児が報告されています。1人目の患者は出生時に顔と首に線状の皮膚欠損が見られましたが、生後数か月でこれらは消失しました。しかし、筋緊張の低下(筋肉が柔らかく力を入れにくい状態)や成長障害があり、生後数か月で心停止を起こして亡くなりました。2人目の患者では、出生前の超音波検査で脳の異常が確認され、出生後2か月でけいれん発作を発症しました。この患者は心筋症のため生後6か月で心臓移植を受け、その後も発達の遅れや運動機能の障害が続きました。
この疾患の症状は非常に多様で、患者ごとに大きく異なります。ある患者では皮膚の異常が顕著である一方、別の患者では目や神経系の異常が目立つ場合があります。また、軽症の親が重症の子どもを持つケースもあるため、この疾患の重症度を事前に予測することは非常に難しいとされています。

治療は、症状に応じた対症療法が中心です。例えば、皮膚の異常には皮膚科医によるケアが、けいれん発作や神経系の異常には小児神経科医の対応が必要です。心筋症がある場合には、心臓専門医による継続的な診察と治療が行われます。また、早期診断のためには遺伝子検査が非常に重要です。特にNDUFB11の変異を確認することで、確定診断が可能となります。
LSDMCA3はこれまでに報告されている症例が非常に少なく、研究が進められている段階です。しかし、この疾患を理解することは、発達障害や他の遺伝性疾患におけるミトコンドリアの役割を明らかにする重要な鍵となるでしょう。さらに、稀少疾患であっても、適切な診断と治療を行うことで患者の生活の質を向上させる可能性があります。このような疾患について医療者や一般の人々が知識を深めることは、患者とその家族の支援に大いに役立つと考えられます。
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
[5_UBA1] 脊髄性筋萎縮症X連鎖2(Spinal muscular atrophy X-linked 2 (SMAX2))

脊髄性筋萎縮症X連鎖型2型(Spinal muscular atrophy X-linked 2, SMAX2)は、非常に稀な神経筋疾患で、X染色体上のXp11.3領域に位置するUBA1遺伝子の変異によって引き起こされます。この病気は主に乳児期に発症し、脊髄前角細胞と呼ばれる運動ニューロンが徐々に損傷を受け、結果として筋力低下や筋萎縮を伴う進行性の症状が現れます。さらに、この疾患は早期の重度な呼吸器合併症を伴うことが多く、生存期間が短いことが特徴です。
UBA1遺伝子は「ユビキチン様修飾因子活性化酵素1」(Ubiquitin-like modifier-activating enzyme 1)をコードしており、この酵素はユビキチン-プロテアソーム系と呼ばれる細胞内のタンパク質分解経路の最初のステップを担っています。この経路は、細胞内で不要になったり損傷したタンパク質を分解するシステムであり、細胞の正常な機能維持に不可欠です。UBA1酵素は、ユビキチンという小さなタンパク質を活性化し、特定の酵素(E1酵素)と結合させることで、タンパク質分解のプロセスを開始します。この過程は、単にタンパク質の分解に留まらず、DNA修復や細胞分裂ストレスへの応答、さらにはDNA損傷部位への特定のタンパク質の集積にも関わっています。UBA1の機能喪失により、これらの重要なプロセスが妨げられ、細胞レベルでの異常が蓄積することで、SMAX2のような深刻な疾患が発症すると考えられています。

SMAX2の臨床症状は、生まれた直後から観察されることが多く、主に筋肉の低緊張(筋緊張が低い状態)、反射消失、そして先天性の関節拘縮が挙げられます。この病気の最も大きな課題の一つは、呼吸筋が影響を受けることで呼吸不全が進行する点です。そのため、早期の呼吸管理が極めて重要です。
この病気の診断は、臨床的な症状と分子遺伝学的検査の両方に基づいて行われます。具体的には、UBA1遺伝子の変異が男性患者で確認されることで確定診断となります。SMAX2はX連鎖型遺伝形式を持つため、変異遺伝子を持つ女性は通常無症状の保因者となりますが、次世代に50%の確率で変異を伝える可能性があります。

患者の管理には、神経内科、整形外科、呼吸器科、栄養療法専門家など、多分野の医療チームによる継続的なケアが求められます。呼吸器管理では、非侵襲的な人工呼吸器(BiPAPなど)の使用が推奨され、必要に応じて気管切開が検討されます。また、栄養補給のための胃瘻(いろう)造設や、便秘の管理も重要です。進行性の関節拘縮や脊柱側弯症に対しては、理学療法や作業療法、場合によっては外科的治療が行われます。
興味深いことに、SMAX2は従来の脊髄性筋萎縮症(Spinal muscular atrophy, SMA)とは異なる病態を示すことが報告されています。具体的には、運動ニューロンだけでなく感覚神経系も影響を受け、小脳の発達や退行異常が認められるケースもあります。このため、一部の専門家は、SMAX2を「運動・感覚ニューロパチー」として分類すべきだと考えています。

また、SMAX2の患者では、呼吸器の合併症が乳幼児期の死亡の主な原因となっています。しかし、侵襲的治療を行った例では、まれに10代まで生存したケースも報告されています。このような場合には、治療の倫理的側面や患者・家族の意思決定を尊重することが非常に重要です。
最後に、SMAX2の病態は、UBA1遺伝子変異によるユビキチン-プロテアソーム系の異常が神経発達にも影響を及ぼす可能性を示唆しています。この知見は、SMAX2のような希少疾患の治療法開発につながるだけでなく、細胞内のタンパク質分解メカニズムのさらなる理解を深める手がかりともなり得るでしょう。
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
引用文献
- Baumbach-Reardon L, Hunter JM, Ahearn ME, et al. Spinal Muscular Atrophy, X-Linked Infantile. 2008 Oct 30 [Updated 2024 May 9]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK2594/
- Dlamini, N., Josifova, D. J., Paine, S. M., Wraige, E., Pitt, M., Murphy, A. J., King, A., Buk, S., Smith, F., Abbs, S., Sewry, C., Jacques, T. S., & Jungbluth, H. (2013). Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscular disorders : NMD, 23(5), 391–398. https://doi.org/10.1016/j.nmd.2013.02.001
- Indrieri, A., & Franco, B. (2021). Linear skin defects with multiple congenital anomalies (Lsdmca): An unconventional mitochondrial disorder. Genes, 12(2), 263. https://doi.org/10.3390/genes12020263
- Morleo M, Franco B. Microphthalmia with Linear Skin Defects Syndrome. 2009 Jun 18 [Updated 2018 Jul 26]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK7041/
- Hamel, C. (2006). Retinitis pigmentosa. Orphanet Journal of Rare Diseases, 1(1), 40. https://doi.org/10.1186/1750-1172-1-40
- Adam MP, Hudgins L, Hannibal M. Kabuki Syndrome. 2011 Sep 1 [Updated 2024 Apr 25]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK62111/
- Scruggs BA, Reding MQ, Schimmenti LA. NDP-Related Retinopathies. 1999 Jul 30 [Updated 2023 Mar 23]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2025. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1331/
- Perez, G., Barber, G. P., Benet-Pages, A., Casper, J., Clawson, H., Diekhans, M., Fischer, C., Gonzalez, J. N., Hinrichs, A. S., Lee, C. M., Nassar, L. R., Raney, B. J., Speir, M. L., van Baren, M. J., Vaske, C. J., Haussler, D., Kent, W. J., & Haeussler, M. (2024). The UCSC Genome Browser database: 2025 update. Nucleic Acids Research, gkae974. https://doi.org/10.1093/nar/gkae974
- Harrison, P. W., Amode, M. R., Austine-Orimoloye, O., Azov, A. G., Barba, M., Barnes, I., Becker, A., Bennett, R., Berry, A., Bhai, J., Bhurji, S. K., Boddu, S., Branco Lins, P. R., Brooks, L., Budhanuru Ramaraju, S., Campbell, L. I., Carbajo Martinez, M., Charkhchi, M., Chougule, K., … Yates, A. D. (2024). Ensembl 2024. Nucleic Acids Research, 52(D1), D891–D899. https://doi.org/10.1093/nar/gkad1049
妊娠中のリスク管理に
NIPTで安心を
ご予約はこちら
Xp11.3 欠失症候群に関する包括的な情報を提供。関連する遺伝子変異や疾患、症状、診断方法、治療アプローチについて詳しく解説します。
NIPT(新型出生前診断)について詳しく見る
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。