

阿尔布莱特综合征样掌骨和跖骨缩短症是一种遗传性疾病,由于部分染色体缺失而导致身体各部位出现异常。 本文根据该病的病例报告,介绍了该病的病因、症状和诊断方法。

ダウン症の検査
気になる費用はこちら
概要
2q37缺失综合症有各种症状和其他表现,取决于负责的基因。奥尔布赖特综合症样的掌骨和跖骨缩短是一个典型的症状。
奥尔布赖特综合症样掌骨和跖骨缩短是一种由2号染色体长臂末端(2q37.3)部分缺失引起的遗传性疾病。是一种极为罕见的疾病,1989年报告了第一例,至今全世界至少有115例。大约一半的受影响者有手指和脚趾短的生理特症,特别是第四个手指据说异常地短。其他身体特症,如特症性的面部特症和稀疏的头发也会出现。神经系统的异常还可能包括运动技能的发育迟缓,如站立、坐立和行走,这些儿童中约有25%有自闭症谱系障碍,社会交流和社会互动受损。其他情况包括炎症性皮肤病,大脑、心脏、消化系统、肾脏和生殖器官的畸形,以及在非常罕见的情况下,一种极其罕见的肝癌–Wilms肿瘤。由于这些特症,该病有时被称为2q37全常染色体全区部分缺失病、奥尔布赖特遗传性骨营养不良样综合症或上腕神经性精神障碍综合症。
原因
奥尔布赖特综合症样的掌骨和跖骨缩短是由染色体的一个区域的缺陷引起的,但目前还不知道为什么会出现这种缺陷。因为大多数受影响的父母(大约95%)具有正常的核型,来自父母的遗传是罕见的,大多数病例是由de novo变异(*1)引起的。因此,它被认为是在父母的精子和卵细胞形成过程中或在胚胎发育的早期阶段,受精卵中的DNA复制错误造成的缺陷。由于总体病例数较少,而且由于de novo变异,父母、兄弟姐妹的基因分型相似的患者也不多,因此分子生物学和遗传学研究不太深入,genotype-phenotype的关联也很少。

发生缺陷的区域往往靠近2号染色体长臂的末端,该缺失已被确定为2q37区域,该区域的纯粹缺失被认为是导致有关疾病的原因。缺失的大小因人而异,已知的范围是2 Mbp~9 Mbp。该区域约有100个基因编码,其中一个或多个基因被认为受到功能受损的影响而发病。最重要的基因之一是HDAC4基因(*2)该基因在一个等位基因中存在缺陷,并伴有杂合子丢失。HDAC4是一种编码组蛋白脱乙酰酶的基因,已知参与骨骼和肌肉发育。奥尔布赖特综合症的掌骨和跖骨缩短与分子水平的关联仍不清楚。
症状
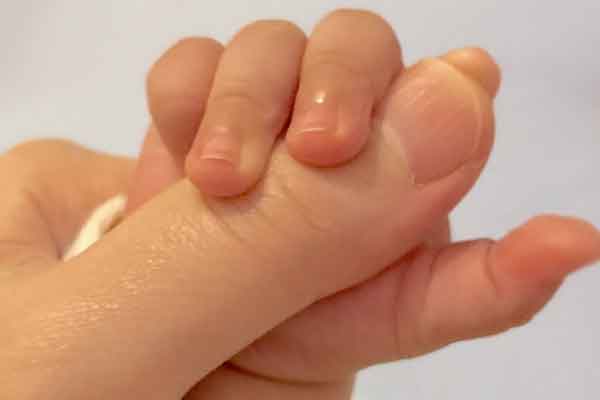
受奥尔布赖特综合症影响的患者,其典型症状主要是身体特症和神经系统异常。身体特症:大约一半的患者有短指症或多指症,特别是手和脚的第4指。此外,大多数病例都有特症性的面部特症,如突出的额头、圆脸、细而拱的眉毛、凹陷的鼻梁、缺失或隆起的鼻腔、薄嘴唇、薄眼皮、上眼睑皱襞、上颚裂、平滑的腭、突出的下颧骨和耳朵异常。此外,还经常出现身材矮小、肥胖、头发稀疏、乳头内陷等乳头异常等症状。还观察到器官发育不良,三分之一的受影响个体出现心脏、消化和肾脏异常。在一些患儿中可能会看到Wilms肿瘤・肾发育不良・支气管软化症,但只有在缺失保持在2q37.1附近时才会出现这种情况。奥尔布赖特遗传性骨营养不良症(*3)的表型见于2q37.3端粒缺失的患者,这些患者后来发展为癫痫发作和囊性肾病。
神经系统异常:观察到发育迟缓和精神区的症状,这些症状在不同的个体中从轻微到严重不等,其中最典型的是由于缺氧症导致的肌肉张力低下。大约一半的病人会有这些症状,据说这些症状会随着时间的推移而改善。此外,它还具有癫痫等癫痫发作症状的高风险特征。此外,还有运动发育延迟和行为异常的报道,如站立、坐立和行走。2q37.3处发生末端缺失的患者,主要在重复行为、交流和社会交往方面有障碍,并且可能表现出自闭症谱系的典型症状。具体来说,如间歇性攻击、多动症、注意力缺失和强迫症以及睡眠紊乱。人们注意到这些神经发育衍生的异常的严重程度和性状的数量取决于基因缺失大小,但这种关系的主导性还没有确定,因为仍有太少的病例来证实这一点。如上所述,由于奥尔布赖特综合症掌骨和跖骨短缩的治疗法的症状如出一辙,所以基本上是针对上述症状给予对症治疗。此外,还进行对肥胖等继发性疾病的健康管理。
诊断
通过仔细记录上述的特症性异常,在一些新生儿和婴儿中,可以做出类似奥尔布赖特综合症掌骨和跖骨缩短临床诊断的可能。然而,在大多数情况下,诊断是在较晚年龄的儿童中确认的。因此,超声检查和放射检查被用来检测最初的身体发现。由于奥尔布赖特综合症的掌骨和跖骨缩短是一种de novo突变,除了少数情况外,对父母的基因检测没有什么价值。病理诊断是通过检查基因组缺陷进行的,大致可分为细胞遗传学和分子遗传学检查,具体如下。
细胞遗传学检查:在80%的奥尔布赖特综合症掌骨和跖骨缩短的病例中,可以通过染色体检测如简单核型分析(*4)确认诊断。然而,这种方法仅限于相对较大的缺陷区域。由于缺失区较小的患者在正常染色体检测中核型正常,因此需要结合下述分子遗传学检测,才能对缺失区个体差异较大的疾病进行高精度、高灵敏度的诊断。
分子基因检测:使用分子生物学技术进行基因诊断。主要使用FISH(Fluorescent in situhybridization)法、DNA微阵列法。
FISH法
这是与目标基因区域互补的单链DNA被荧光标记,并使用荧光显微镜观察的一种技术。荧光标记的单链DNA称为FISH探针,可以根据目的基因区域的序列进行任意设计。由于奥尔布赖特综合症样的掌骨和跖骨缩短常常显示出染色体末端区域的缺陷,所以经常设计针对端粒和副端粒区域的FISH探针,有些探针已经作为试剂盒在市场上销售。在这些多数重复的区域,FISH探针以多个数值结合,这增加了荧光色素的有效浓度并增强了信号。另一方面,如果发生了缺失,FISH探针就不能与目标区域结合,并被随后的洗涤操作洗掉,因此就检测不到荧光信号。因此,用DAPI或Hoechst等与DNA特异性结合的染料染色染色体并与FISH探针杂交,可以绘制出目标区域存在于染色体上的哪个区域。FISH方法是一种可以检测极小区域缺损的技术,可以高精度诊断奥尔布赖特综合症掌骨和跖骨缩短。但是,缺失区域必须包含FISH探针的目标序列。在某些情况下,亚端粒区的中间部分已经缺失,在这种情况下,如果将FISH探针设计在端粒区,会导致假阴性而无法检测到,所以FISH的选择和设计探头极其重要。
DNA微阵列
使用将已知DNA片段(在这种情况下是整个人类基因组的片段)密集放置在基板上的芯片,将从待检查的组织中提取的DNA与作为对照的具有正常序列的DNA杂交和比较的方法。如果样品中含有基质上的互补DNA片段,它们会与基质上的互补序列结合并发出荧光。通过比较芯片上的荧光信号与对照的空间分布,有可能对基因组中哪里发生了缺陷进行全面分析。这样的方法具体称为CGH(Comparative genomic hybridization)微阵列。该方法分析染色体上基因位点拷贝数的变化,虽然拷贝数增加可以以高灵敏度检测,而部分缺失在5~10 Mbp的大小以下难以检测到。
未来展望
近年来,通过新一代测序仪(next-generation-sequencing)基因组序列分析得到了发展,预计将与genotype-phenotype的关联高度相关。然而,由于设备本身的成本和运行成本,将这种方法用于实际诊断应用还不现实。上面提到的两种方法是相对便宜的,因为它们是基于荧光检测的原理,这些方法实际上是用于病理诊断的应用。目前,以相对较低的成本进行外包已成为可能,而新一代测序仪设备本身的价格也在逐步下降,在试剂等方面,我们正在设计通过以协同方式同时分析多个样本来减少运行次数来降低运行成本的方法。
今后genotype-phenotype关联进一步发展,则可以根据缺失位置和大小预测会出现什么样的症状。这不仅有助于阐明奥尔布赖特综合症掌骨和跖骨缩短的原因和开发治疗方法,而且有助于阐明其他重要的遗传性疾病。特别是2q37.3区域被认为包含与自闭症谱系障碍发病相关的候选基因,可以说是一个重要的基因组区域。240.1-242.2 Mb的最小缺失间隔可导致自闭症谱系障碍、过度运动行为和癫痫,并且包含突触小泡的轴突转运蛋白等至少29个基因。有趣的是这个2.1 Mb的缺失区间与湿疹的最小缺失区间(1.5 Mb)相重合。近年来,有人指出自闭症谱系障碍与特应性皮炎之间存在关联,尽管这尚未得到证实,但从缺失间隔的重合genotype-phenotype的观点来看似乎合理。因此,由于2q37区域的缺失已被观察到有不同的表现,进行更详细的分析,可能会发现疾病之间意想不到的关联。如果这样的话,我们不仅可以期待开发全新的治疗方法,还可以进一步期待现有的repurposing药物和治疗方法。

摘要
本文介绍了奥尔布赖特综合症样掌骨和跖骨缩短,这是一种由2q37缺失的基因组区域缺失引起的遗传疾病。由于是极为罕见的疾病,还有很多未知点,未来将通过积极结合最新的遗传学方法,希望今后能积极结合最新的遗传技术,对该病的机制和基因型genotype-phenotype进行分子生物学分析。这些信息也有望帮助阐明其他遗传疾病的机制、诊断方法和治疗方法。
Hiro诊所NIPT,也提供这种染色体的部分缺失和重复常染色体全领域部分缺失・重复疾病的诊断。请考虑结合全染色体来进行检测。
脚注
- (*1) de novo变异:所谓的突变,不是从父母那里遗传来的突变,而是个体中新出现的突变。也叫新生变异。
- (*2) HDAC4基因:蛋白去甲基化酶(HDAC4)的编码组基因。通过Transforming growth factor β1 (TGF-β1)在诱导纤维细胞向成肌细胞分化中起重要作用。
- (*3) 奥尔布赖特遗传性骨营养不良综合症:特别是当身体和智力发育障碍、短指症、身材矮小、肥胖同时存在时,会这样称呼。
- (*4) 核型分析:使用染色体的特症带模式进行核型分析。包括使用Hoechst色素的Q-band分析和使用Giemsa染色的G-band分析,这可以从染色体结构中简单地确定染色体的特性、倍性和易位等情况。
阿尔布莱特综合征样掌骨和跖骨缩短症是一种遗传性疾病,由于部分染色体缺失而导致身体各部位出现异常。 本文根据该病的病例报告,介绍了该病的病因、症状和诊断方法。
NIPTについて詳しく見る
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。







