
このページのまとめ
染色体の一部が失われる「部分欠失」や、余分に増える「部分重複」は、知的障害や発達の遅れなどにつながることがあります。NIPTy Standardでは、常染色体44本のどの部位の欠失・重複も調べられます。ただしNIPTは非確定的検査(スクリーニング)です。陽性となった場合は、羊水検査で確定診断を行います。ヒロクリニックでは、東京衛生検査所(国内)で解析し、最短2〜5日で結果をお届けします。
このページでわかること
- 部分欠失・部分重複とは何か、なぜ症状が出るのか
- NIPTでどこまで調べられるか(対象・限界・確定検査との違い)
- 染色体ごとの代表的な疾患と報告例(1番〜22番)
- 陽性だったときの進め方と、羊水検査サポート費用の補助
- プランの選び方と、結果までの日数・相談窓口
監修:岡 博史(ヒロクリニック統括院長)/NIPTは非確定的検査であり、確定診断には羊水検査が必要です。
はじめに知っておきたい用語
- 部分欠失(けっしつ)
- 染色体の一部分が失われている状態です。そこにある遺伝子の働きが失われ、発達や身体の形成に影響することがあります。
- 部分重複(ちょうふく)
- 染色体の一部分が余分に増えている状態です。遺伝子が過剰に働くことで症状が出ることがあります。
- Mb(メガベース)
- DNAの長さの単位です。1Mbは約100万塩基。東京衛生検査所の解析では、500万塩基(0.23%)以上の欠失・重複を報告対象としています。
- de novo(デノボ)変異
- 両親にはなく、赤ちゃんに新しく生じた変化を指します。
- 意義不明変異(VUS)
- 欠失や重複が見つかっても、病気との関連がまだはっきりしないものを指します。
- 顕性(けんせい)・潜性(せんせい)
- 従来「優性・劣性」と呼ばれていた遺伝の伝わり方です。片方の変化で症状が出るものを顕性、両方そろって出るものを潜性といいます。
検査を受ける前に:NIPT(新型出生前診断)は、対象となる染色体の変化について可能性を調べる非確定的検査(スクリーニング)です。陽性という結果はそのまま診断ではありません。確定診断には羊水検査が必要です。ヒロクリニックでは、他院での羊水検査にも使える費用サポート(最大10万〜30万円の補助)と、医師による遺伝カウンセリングをご用意しています。
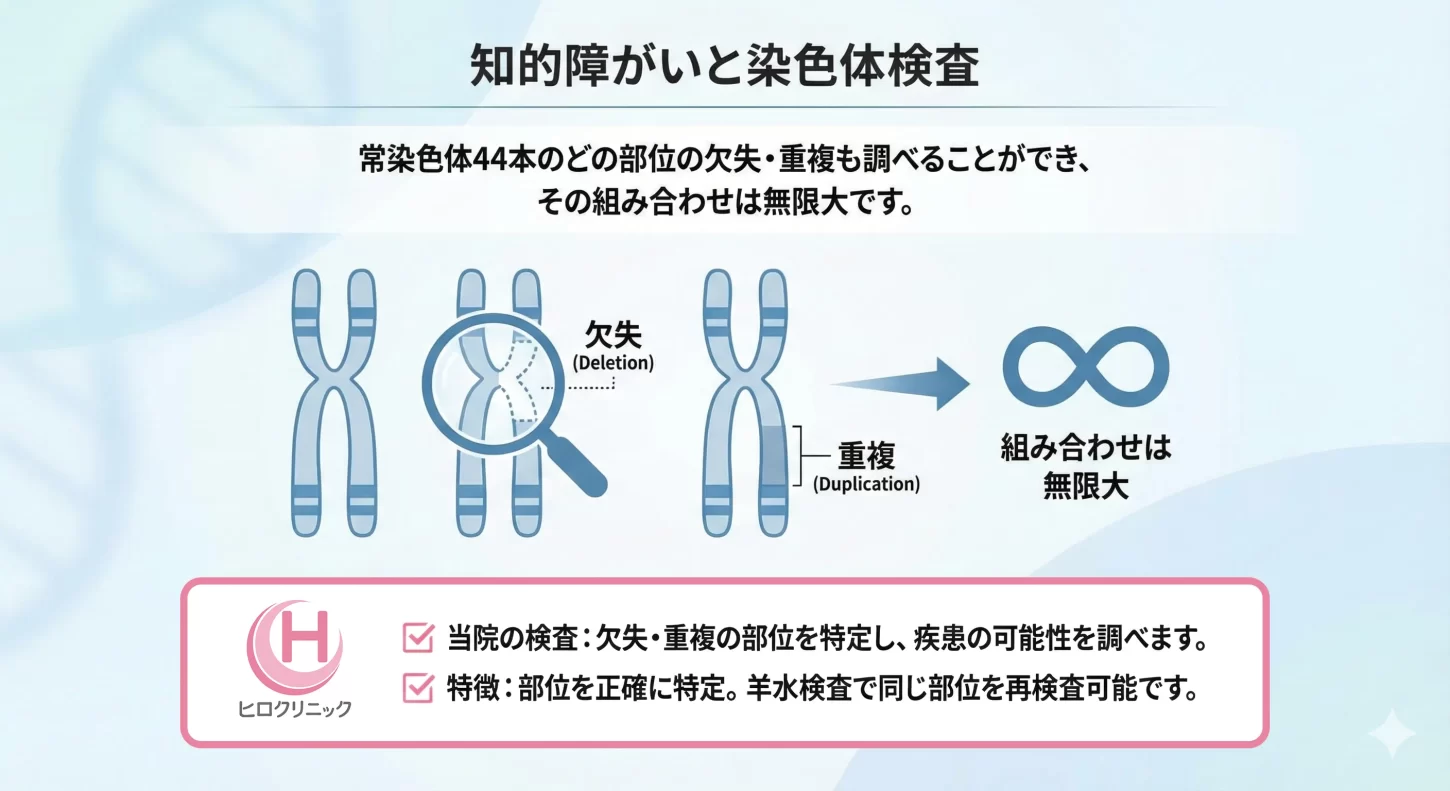

ヒロクリニックが検査を依頼している東京衛生検査所の次世代シーケンサーでは、500万塩基以上の欠失・重複が検出された場合に、検査結果として報告しています。
微小欠失疾患は、100万塩基から500万塩基程度の欠失でも症状を発症することがありますが、症例によっては500万塩基以上の欠損を伴うこともあります。疾患特有の遺伝子欠損を超える大きな欠失があった場合でも、その遺伝子が欠損しているため、同様の症状やより重度の疾患を引き起こす可能性があります。このため、欠損部位が疾患特有の部位に含まれている場合、欠損範囲が広くても、同じ診断名を付けることが妥当だと考えています。微小欠失と呼ぶと誤解を招く恐れがあるため、当サイトでは「部分欠失疾患」として記載しています。
欠失・重複は、その部位に存在する遺伝子がどのような働きを持っているかによって、さまざまな症状を引き起こします。例えば、ある酵素を作り出す遺伝子を含む染色体が欠損すると酵素欠損症が発生し、骨形成に関わる遺伝子を含む染色体が重複すると骨形成異常が見られます。
従来、染色体の欠失・重複は、染色体を染色し顕微鏡下で検出していましたが、この方法では1,000万塩基(10Mb)以上の欠失・重複しか検出できませんでした。近年、次世代シーケンサーやマイクロアレイ染色体検査を用いることで、より精度の高い検査が実施できるようになり、欠失・重複は特定の染色体に限定されず、全染色体にわたって発生することが明らかになりました。これにより、欠失・重複と、それに伴う臨床的所見との関連が報告され、異常の認められた遺伝情報がデータベース化されています。このデータベースには、共通の異常領域と特徴的な所見に基づく複数の報告例が含まれており、稀な例や新たに報告されたケースもあります。
したがって、独立した疾患として報告されていない欠失・重複がある場合、症例報告やゲノムデータベースを参照して症状などを調査する必要があります。東京衛生検査所では、染色体の部分的な欠失・重複が検出された際に、国内外の信頼性の高いデータベースを検索し、該当する疾患情報を担当医師に提供しています。
すべての重複領域における遺伝子と臨床所見との関連性は、まだ明確にはなっていません。また、欠失・重複が認められた場合でも、症状が現れないことがあります。これは、欠失・重複する部位に生命活動や身体の形成に関わる重要な遺伝子が存在しない場合、または遺伝子が存在していても病的所見を示さない場合があるためです。例えば、8番染色体の短腕(8p23.2)の重複は、250万塩基(2.5Mb)に及ぶものでありながら、がん抑制遺伝子のみが含まれているため、異常所見は認められず、正常変異として理解されることもあります。
欠失・重複が認められた場合、その病的な影響を鑑別する必要がありますが、これまでに報告されたことのないケースについては、その判断が難しいことや、欠失・重複との関連が不明な場合(variant of unknown significance: VUS = 意義不明変異)があることをご理解ください。
名もなき症例
全染色体部分欠失・重複疾患で近年になって報告された症例の論文の一例です。
| 染色体 | 染色体座位 | 論文 |
|---|---|---|
| 2番 | 2p16.3-p21 | 2番染色体(2p16.3‐p21)の隣接遺伝子欠失:リンチ症候群の原因として(翻訳) |
| 7番 | 7q21.3-q31.1 | 7番染色体(7q21.3-q 31.1)部分重複: まれな分節性ゲノム異数性: 症例報告と遠位および類似の分節が関与した症例のレビュー(翻訳) |
| 12番 | 12q24.31-q24.33 | 12番染色体(12q24.31-q24.33)部分欠失に伴う自閉症:非常に稀な疾患の追加報告(翻訳) |
| 14番 | 14q | 14番染色体の部分重複(翻訳) |
| 19番 | 19p13.13 | 19番染色体(19p13.13)の新規微小欠失/微小重複症候群(翻訳) |
全染色体部分欠失・重複疾患の報告例
ここからは、1番染色体から22番染色体まで、部分欠失・部分重複でみられる代表的な疾患と報告例をまとめています。各見出しをタップすると詳しい説明が開きます。専門的な内容が続きますので、気になる染色体からご覧ください。いずれもNIPTで可能性を調べられますが、確定には羊水検査が必要です。
| 染色体 | 欠失部 | 症候群 | 備考 |
|---|---|---|---|
| 1番 | 1p12 | アラジール症候群(Alagille症候群) | |
| 1q21.1 | 1q21.1微細欠失症候群 | ||
| 1p36 | 1p36欠失症候群 | 発生頻度(出生時)4,000〜10,000件中1件 成長障害、重度精神発達遅滞、難治性てんかんなどの症状 |
|
| 2番 | 2q13 | ネフロン癆(Nephronophthisis)1型 | |
| 2p21 | 全前脳胞症 | ||
| 2q37.3 | オルブライト(Albright)症候群様中手骨・中足骨短縮 | ||
| 3番 | 3q29 | 3q29微細欠失症候群 | |
| 4番 | 4p16.3 | ウォルフヒルシュホーン症候群(Wolf-Hirschhorn症候群) | 発生頻度(出生時)50,000件中1件 重度の精神発達の遅れ、成長障害、難治性てんかん、多発形態異常。 |
| 5番 | 5p13.2 | コルネリア・デ・ランゲ症候群(Cornelia de Lange症候群) | |
| 5p15.2 | 猫鳴き症候群 | 発生頻度(出生時)20,000〜50,000件中1件 低出生体重、成長障害、甲高い猫のなき声のような啼泣。顔貌所見や筋緊張低下、精神運動発達の遅れ。 5番染色体部分欠失:ねこなき症候群における高解像度マッピング |
|
| 5q35.3 | ソトス症候群(Sotos症候群) | 欠失型と重複型とでは一部症状の差異が指摘されている。 発生頻度(出生時)14,000件中1件 |
|
| 7番 | 7q11.23 | ウィリアムズ症候群(Williams症候群) | |
| 7p13 | パリスター・ホール症候群(Pallister-Hall症候群) | ||
| 7p14.1 | グレイグ脳多合指趾症(Greig 脳多合指趾症) | ||
| 7p21.1 | セートレ・ヒョッツェン症候群(Saethre-Chotzen症候群) | ||
| 7q36.3 | 全前脳胞症3型 | ||
| 8番 | 8q12.2 | チャージ症候群(CHARGE症候群) | |
| 8p23.1 | 8p23.1微細欠失症候群 | ||
| 8q23.3 | 毛髪・鼻・指節症候群1型 | ||
| 8q24.11 | ランガー・ギデオン症候群(Langer-Giedion症候群) | ||
| 11番 | 11p11.2 | ポトツキ・シェファー症候群(Potocki-Shaffer症候群) | |
| 11p13 | WAGR症候群 | ||
| 12番 | 12q24.13 | ヌーナン症候群(Noonan症候群) | RAF1において重複の報告1例、欠失の報告1例 |
| 13番 | 13q14.2 | 網膜芽細胞腫・発達遅滞 | |
| 13q32.3 | 全前脳胞症5型 | ||
| 15番 | 15q11.2〜q13 | プラダー・ウィリー症候群(Prader-Willi症候群) | 父親由来の遺伝子の欠失で、母親由来の遺伝子に起因する 発生頻度(出生時)10,000〜25,000件中1件 筋緊張低下、色素低下、外性器低形成。 |
| 15q11.2〜q13 | アンジェルマン症候群(Angelman症候群) | UBE3Aの機能喪失により発症 発生頻度(出生時)12,000件中1件 重度の精神発達の遅れ、てんかん、失調性運動障害、行動異常、睡眠障害、低色素症、特徴的な顔貌。 |
|
| 16番 | 16p11.2 | 16p11.2微細欠失 | |
| 16p13.11 | 16p13.1微細欠失 | ||
| 16p13.3 | ルビンシュタイン・テイビ症候群(Rubinstein-Taybi症候群) | 発生頻度(出生時)125,000件中1件 | |
| 17番 | 17p13.3 | ミラー・ディカー症候群(Miller-Dieker症候群) | |
| 17p11.2 | スミス・マギニス症候群(Smith-Magenis症候群) | 発生頻度(出生時)15,000~25,000件中1件 | |
| 17q11.2 | 神経芽細胞種1型 | ||
| 20番 | 20p12.23 | アラジール症候群(Alagille症候群) | |
| 22番 | 22q11.2 | ディ・ジョージ症候群(DiGeorge症候群)2型 22q11.2欠失症候群 |
発生頻度(出生時)4,000件中1件 先天性心疾患、精神発達遅延、特徴的顔貌、免疫低下、口蓋裂・軟口蓋閉鎖不全、鼻声、低カルシウム血症。 |
| 22q13.33 | フェラン・マクダーミド症候群(Phelan-McDermid症候群) |
報告例は部分欠失の一部です
東京衛生検査所で使用している次世代シーケンサーは500万塩基以下の欠失・重複は検出できません。
症候群の全ての症例を検出できるわけではありません。
| 染色体 | 重複部 | 症候群 | 備考 |
|---|---|---|---|
| 1番 | 1q21.1 | 1q21.1微細重複症候群 | 1q21.1 部分重複(翻訳) |
| 2番 | 2p21 | 全前脳胞症 | |
| 3番 | 3q29 | 3q29微細重複症候群 | |
| 5番 | 5p13.2 | コルネリア・デ・ランゲ症候群(Cornelia de Lange症候群) | |
| 5q35.3 | ソトス症候群(Sotos症候群) | 欠失型と重複型とでは一部症状の差異が指摘されている。 | |
| 8番 | 8p23.1 | 8p23.1微細重複症候群 | |
| 9番 | 9q34.13 | 結節性硬化症1型 | TSC1遺伝子が原因 5,800件中1人 |
| 10番 | 10q24.3 | 染色体10q24重複症候群 | 11番 | 11p13 | WAGR症候群 |
| 12番 | 12q24.1 | ヌーナン症候群(Noonan症候群) | RAF1において重複の報告1例、欠失の報告1例 |
| 13番 | 13q32.3 | 全前脳胞症5型 | |
| 15番 | 15q26qter | 過成長・知的障碍 | |
| 16番 | 16p11.2 | 16p11.2微細重複 | |
| 16p13.3 | 結節性硬化症2型 | TSC2遺伝子が原因 5,800件中1人 |
|
| 16p13.3 | ルビンシュタイン・テイビ症候群(Rubinstein-Taybi症候群) | CREBBP遺伝子が原因 発生頻度(出生時)125,000件中1件 |
|
| 16p13.11 | 16p13.1微細重複 | ||
| 17番 | 17p11.2 | ポトツキ・ルプスキ症候群(Potocki-Lupski症候群) | 17番染色体(17p11.2)部分重複:発達遅延のある小児における重複 |
| 17p12 | シャルコー・マリー・トゥース病(Charcot-Marie-Tooth)1A型 | ||
| 17q21.31b | 17q21.31微細重複症候群 | ||
| 22番 | 22q11.1 | 猫の目症候群 | |
| 22q11.2 | 22q11.2重複症候群 | 先天性心疾患、精神発達遅延、特徴的顔貌、免疫低下、口蓋裂・軟口蓋閉鎖不全、鼻声、低カルシウム血症。 |
報告例は部分欠失・重複の一部です
東京衛生検査所で使用している次世代シーケンサーは500万塩基以下の欠失・重複は検出できません。
症候群の全ての症例を検出できるわけではありません。
染色体1
部分欠失では、染色体の特定領域に存在する遺伝子が失われ、その結果、発達障害や特定の症候群が引き起こされることがあります。
- 原因: 1番染色体の短腕(1p36)に欠失があることで発生します。
- 症状: 知的障害、発達遅延、筋力低下(低緊張)、行動問題、心臓の異常、顔面の特徴的な異常(例えば、広い前額、浅い目など)が一般的です。その他、けいれん発作や聴覚の問題も伴うことがあります。
1q欠失症候群
- 原因: 1番染色体の長腕(1q)の欠失が原因で発生します。
- 症状: 知的障害や発達遅延、顔の異常、心臓や腎臓の異常が見られることがあります。また、筋力低下やてんかんも報告されています。
1q21.1欠失症候群:1番染色体の長腕(q)の21.1領域が欠失している場合、心臓疾患、精神発達遅延、自閉症スペクトラム障害などのリスクが高まります。
1q42.13欠失症候群(1番染色体の長腕の42.13領域の欠失による症候群)
部分重複では、染色体の特定領域に遺伝子が過剰に存在し、健康や発達に異常が現れます。
1p36重複症候群
- 原因: 1p36領域の遺伝子が重複することにより、遺伝子の過剰発現が引き起こされます。
- 症状: 重複の位置や範囲によって異なりますが、発達遅延、知的障害、特徴的な顔の形態異常が見られます。心臓の異常や筋力低下、行動上の問題(自閉症傾向など)が報告されています。
1q重複症候群
- 原因: 1番染色体の長腕(1q)が重複することで、特定の遺伝子が過剰に存在します。
- 症状: 知的障害、発達遅延、身体的な異常(心臓や腎臓の異常など)や筋力低下が見られることがあります。重複の位置に応じて、症状の重さが異なる場合があります。
染色体2
染色体3
染色体4
4番染色体の部分欠失は、染色体の一部が欠失し、その結果、発達障害や身体的異常が発生することがあります。
4p欠失症候群(Wolf-Hirschhorn症候群)
- 原因: 4番染色体の短腕(4p)の一部が欠失することによって発症します。
- 症状: 重度の知的障害、発達遅延、低身長、特徴的な顔の形態(「兜型の顔」など)、けいれん、心臓の奇形、筋力低下などが見られます。この症候群は珍しい遺伝的疾患として知られています。
4q欠失症候群
- 原因: 4番染色体の長腕(4q)の一部が欠失していることが原因です。
- 症状: 発達遅延、知的障害、心臓や腎臓の異常、顔の形態異常が報告されています。また、筋力低下や運動発達の遅れも見られることがあります。
5番染色体
部分欠失は、染色体の一部が欠失することで、重要な遺伝子が機能しなくなり、健康や発達に悪影響を及ぼすことがあります。
5p欠失症候群(クライ・デュ・シャット症候群)
- 原因: 5番染色体の短腕(5p)の一部が欠失することによって発症します。この症候群は「猫鳴き症候群」とも呼ばれ、特徴的な「猫の鳴き声」に似た泣き声が新生児期に見られることが多いです。
- 症状: 知的障害、発達遅延、筋力低下、特徴的な顔つき(小さな顎、広い鼻、下がった目)、低体重、行動問題が含まれます。重度の症例では、心臓の奇形や呼吸器系の問題が見られることもあります。
5q欠失症候群
- 原因: 5番染色体の長腕(5q)が欠失することで引き起こされる血液疾患です。この異常は骨髄異形成症候群(MDS)と関連し、赤血球を作る細胞に異常が見られることがあります。
- 症状: 貧血、骨髄不全、疲労感、免疫系の異常が報告されています。5q欠失は特に成人女性に多く見られる傾向があります。
6番染色体
7番染色体
7番染色体の部分欠失は、遺伝子の一部が失われることで、健康上の問題を引き起こします。特に以下の症候群がよく知られています。
ウィリアムズ症候群(Williams Syndrome)
- 原因: 7番染色体の長腕(7q11.23)にあるELN(エラスチン)遺伝子の欠失が主な原因です。
- 症状: 知的障害、心臓血管の問題(特に大動脈弁の狭窄)、特徴的な顔つき(小さな鼻、広い口)、社交的な性格が見られます。さらに、発達遅延や学習障害も伴うことがあります。
7q欠失症候群
- 原因: 7番染色体の長腕(q)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、顔面の異常(目の間が広いなど)、成長障害、心臓や腎臓の問題が報告されています。
7番染色体の部分重複は、特定の遺伝子が過剰に存在することで、健康や発達に影響を与えます。以下のような症状が見られる場合があります。
7q11.23重複症候群
- 原因: 7番染色体の同じ領域(7q11.23)が重複することで発症します。ウィリアムズ症候群と逆の状態です。
- 症状: 自閉症スペクトラム障害(ASD)、発達遅延、言語の遅れ、学習障害が主な特徴です。行動問題や感覚過敏も報告されています。
7p重複症候群
- 原因: 7番染色体の短腕(p)が重複することで引き起こされます。
- 症状: 発達遅延、知的障害、特徴的な顔貌、心臓や消化器の問題が見られることがあります。行動障害や睡眠障害も報告されています。
8番染色体
8番染色体の部分欠失は、染色体の一部が失われることにより、発達障害や身体的な異常を引き起こします。以下は、8番染色体の欠失に関連する代表的な症候群です。
8p欠失症候群(8p微細欠失症候群)
- 原因: 8番染色体の短腕(8p)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、特徴的な顔貌、先天性心疾患、運動協調性の問題が見られます。言語発達の遅れや行動上の問題も報告されています。
ラングラー・グルーベル症候群(Langer-Giedion症候群)
- 原因: 8番染色体の長腕(8q)にあるTRPS1遺伝子とEXT1遺伝子の欠失によって引き起こされます。
- 症状: 知的障害、特徴的な顔つき、骨格の異常(特に指や骨の成長障害)、脱毛症(毛髪の成長不全)などが見られます。
9番染色体
9番染色体の部分欠失は、染色体の一部が失われることで発達障害や身体的異常を引き起こします。以下のような症候群が知られています。
9p欠失症候群
- 原因: 9番染色体の短腕(9p)の一部が欠失することにより発症します。
- 症状: 知的障害、発達遅延、特徴的な顔つき(広い鼻、上向きの鼻孔、離れた目など)、成長障害、運動機能の遅れが見られることがあります。心臓や腎臓の異常も報告されています。
- 別名: アルフ・フィース症候群(Alfi’s Syndrome)とも呼ばれます。
9q欠失症候群
- 原因: 9番染色体の長腕(9q)の一部が欠失することで発生します。
- 症状: 知的障害、発達の遅れ、筋力低下、心臓や消化器系の異常、免疫不全などが見られます。欠失した領域に応じて症状の重さが異なります。
10番染色体
10番染色体の部分欠失は、染色体の一部が失われることで、発達障害や身体的な異常を引き起こすことがあります。
10q欠失症候群(10q微細欠失症候群)
- 原因: 10番染色体の長腕(10q)の一部が欠失することで発症します。
- 症状: 知的障害、発達遅延、特徴的な顔貌、心臓や腎臓の異常が見られることがあります。また、運動機能の遅れや言語の発達遅延も一般的です。
- 治療: 欠失症候群は、早期の介入と療育が重要であり、発達支援や医療的ケアが必要です。心臓の異常などには外科的治療が行われる場合もあります。
10p欠失症候群
- 原因: 10番染色体の短腕(10p)の一部が欠失することで発症します。
- 症状: 発達遅延、知的障害、成長障害、特徴的な顔つき、心臓や腎臓の異常が見られることがあります。顔の特徴的な形態には、広い鼻、上向きの鼻孔、異常に広がった目などが含まれます。
染色体11
11番染色体の部分欠失は、染色体の一部が失われることで、特定の遺伝子の機能が欠如し、発達遅延や身体的な異常を引き起こすことがあります。以下に代表的な症候群を示します。
ウィルムス腫瘍(WT1遺伝子欠失)
- 原因: 11番染色体の短腕(11p)にあるWT1遺伝子の欠失によって発症します。この遺伝子は腎臓の発達に関与し、腫瘍抑制にも重要な役割を果たします。
- 症状: 腎臓のがん(ウィルムス腫瘍)、泌尿生殖器の異常、場合によっては性分化異常を伴います。
ベックウィズ・ウィーデマン症候群(BWS)
- 原因: 11p15.5領域における遺伝子の欠失または機能不全により発症します。これはIGF2などの成長に関与する遺伝子の異常に関連しています。
- 症状: 巨大児症、臓器肥大、へそヘルニア、腫瘍発生のリスクが高いことが特徴です。
ヤコブセン症候群
- 原因: 11番染色体の長腕(11q)の一部が欠失することで発症します。
- 症状: 知的障害、運動遅延、特徴的な顔つき(広い額、下がった目尻)、出血傾向、心臓の異常などが報告されています。
12番染色体
部分欠失は、染色体の特定の領域が失われていることで、さまざまな遺伝子が不足し、その結果として特定の症状が現れる可能性があります。12番染色体における欠失の影響は、欠失した領域とそこに含まれる遺伝子に依存します。
- 症状の例: 知的障害、発達遅延、身体の異常(心臓や腎臓の奇形など)、低身長や筋力低下など。
12q15欠失症候群のように、特定の領域(12番染色体の長腕)が欠失すると、特有の症状が現れることが報告されています。 - 診断: 欠失は染色体検査(FISHやCGHアレイなど)で特定されます。
主な症候群
- 12q欠失症候群:12番染色体の長腕(q)の一部が欠失している場合を指します。一般的な症状には、発達遅延、知的障害、筋緊張低下、特徴的な顔貌、心臓の異常などがあります。
- 12p欠失症候群:12番染色体の短腕(p)の一部が欠失している場合です。これにより、発達遅延や身体的な異常が生じることがあります。

13番染色体
13番染色体の一部が欠失することで、特定の遺伝子が機能しなくなり、発育や健康に影響を及ぼす可能性があります。
- 13q欠失症候群 13番染色体の長腕(q)の部分が欠失することで発症します。具体的な症状には、知的障害、発育遅延、特定の顔貌異常(目が小さい、鼻が低いなど)、指や爪の発達不良、心臓や腎臓の異常が含まれます。
- 網膜芽細胞腫(Retinoblastoma) 13番染色体のq14領域にあるRB1遺伝子の欠失が、網膜芽細胞腫という小児期の眼のがんの原因となることがあります。
13番染色体の部分が重複すると、余分な遺伝情報が含まれるため、過剰な発現が起こり、健康や発達に異常を引き起こす可能性があります。
- 重複症候群: 13番染色体の部分的な重複は非常に稀ですが、知的障害、発育遅延、頭部や顔面の形態異常(例えば、広い額や小さな顎など)が見られることがあります。重複する領域によって症状の重篤度は異なります。
染色体14
14番染色体の一部が欠失すると、その領域に存在する遺伝子が不足するため、発達障害や身体的異常が生じることがあります。
-
症状の例:
- 知的障害や発育遅延
- 特定の顔貌の異常(顔つきの変化や骨格の異常)
- 心臓や腎臓の奇形
- 免疫不全などの症状が報告されています。
- 14q欠失症候群 特に14番染色体の長腕(q)の欠失が知られており、成長障害、筋肉の低緊張、発達の遅れ、さらにはてんかんなどの神経学的症状が伴うことがあります。
染色体15
15番染色体の部分欠失では、以下のような遺伝的疾患が知られています。
プラダー・ウィリー症候群(PWS)
- 原因: 15番染色体の父親由来の一部(通常は15q11-q13領域)の欠失が原因で発生します。まれに遺伝子の機能不全や欠失が原因です。
- 症状: 新生児期からの筋力低下(低緊張)、食欲異常による肥満、知的障害、性腺機能不全などが特徴です。行動上の問題や睡眠障害も見られることがあります。
アンジェルマン症候群(AS)
- 原因: プラダー・ウィリー症候群と同じ領域(15q11-q13)の欠失ですが、こちらは母親由来の染色体で欠失が発生します。
- 症状: 知的障害、運動の協調性欠如、頻繁な笑いや興奮、てんかん発作が主な特徴です。
16番染色体
17番染色体
17番染色体の部分欠失は、染色体の一部が失われることで、重要な遺伝子が不足し、発育や健康に影響を与える可能性があります。
17p13.3欠失症候群(ミラー・ディーカー症候群)
- 原因: 17番染色体の短腕(p)の13.3領域の欠失によって引き起こされます。
- 症状: 重度の知的障害、てんかん、筋肉の低緊張、頭蓋顔面の異常が特徴です。脳の発達に影響を与え、滑脳症と呼ばれる脳の異常を引き起こすことがあります。
スミス・マゲニス症候群(Smith-Magenis syndrome)
- 原因: 17p11.2領域の欠失が原因で発生します。
- 症状: 知的障害、発達遅延、行動異常(例えば、自傷行為)、睡眠障害、特徴的な顔つきなどが見られます。
染色体18
18番染色体の部分欠失は、染色体の一部が欠失することで、遺伝子が失われ、発達や身体の異常が生じます。
18q欠失症候群(18q-症候群)
- 原因: 18番染色体の長腕(q)の一部が欠失することによって発症します。
- 症状: 発育遅延、知的障害、筋力低下、顔面の特徴的な変形、足の変形(扁平足など)、免疫不全、言語の遅れ、行動障害が見られます。また、思春期以降にホルモンの問題が起こることもあります。
18p欠失症候群(18p-症候群)
- 原因: 18番染色体の短腕(p)が欠失することによって発生します。
- 症状: 成長遅延、運動の発達遅延、筋力低下、軽度から中等度の知的障害が一般的です。顔の形態異常、言語発達の遅れなども報告されています。
19番染色体
20番染色体
21番染色体
22番染色体
22番染色体の部分欠失では、遺伝子が欠失することで、発育障害や身体的異常が現れることがあります。特に、22q11.2の欠失が最も一般的です。
キャットアイ症候群(Cat Eye Syndrome)
- 原因: 22番染色体の一部(短腕と長腕の接合部に近い部分)が欠失または重複することで発症します。
- 症状: 目に特徴的な異常(瞳孔が縦に割れる)、肛門や尿路の異常、心臓の異常、発達遅延が報告されています。
微小欠失・重複
微小欠失Microdeletions: 5025人に1人・重複Duplications: 14286人に1人
最近の研究では、報告されたすべての染色体異常の4.7%が、微小欠失を含む欠失であることが示され、出生10,000人あたり1.99人の有病率が得られた(3)。 重複はさらに少なく、出生10,000人あたり0.7人の有病率を示し、報告された全染色体異常の1.6%に相当する(3)。
https://www.sciencedirect.com/science/article/pii/S0015028216629643常染色体潜性疾患
常染色体潜性の障害
常染色体潜性(AR)疾患は、かなりの遺伝性疾患の一部を構成しており、相当な疾病負担の原因である。新生児1000例で約1.75-5人に影響する(これに対し、常染色体顕性疾患では1000例で1.4人)。
https://www.nature.com/articles/s41525-021-00203-x全染色体の異数性および部分欠失部分重複の検査をする必要はないのか?
認可施設で行われるNIPTは、21・18・13トリソミーを中心とした検査が基本です。一方で、それ以外の染色体の異数性や部分欠失・部分重複も、頻度は高くないものの、妊娠経過に関わることが国内外で報告されています。ヒロクリニックでは、東京衛生検査所(国内)での解析を通じて、これらの染色体の変化についても検査データを蓄積してきました。ここでは、当院で得られた検査結果の一部を、海外で報告されているデータとあわせてご紹介します。なお、NIPTは非確定的検査であり、確定診断には羊水検査が必要です。
海外の情報は上記のサイトから取得してみたいと思います。これは専門家のみが見れるサイトなので、簡単に訳してみたいと思います。(誰でも見れますが、専門用語が多いので難しいです)
以下では、海外の公的なNIPTコンソーシアムで報告されているデータの要点をご紹介します。各国の報告と、ヒロクリニックで蓄積している検査データとの間に大きな差はありません。
公的資金による全国一次非侵襲的出生前スクリーニングの結果
ベルギーは、すべての妊婦を対象とした一次スクリーニングとして NIPT を実施し、全額償還した最初の国です。この出版物では、ベルギーのすべての遺伝センターで構成されるコンソーシアムが、すべての妊婦に一次検査オプションとして拡張 NIPT を提供した 2 年間の臨床経験について報告しています。スクリーニングされた153,575人の妊娠のうち、まれな常染色体トリソミーと部分欠失/重複がそれぞれ0.23%と0.07%の症例で見つかりました。侵襲的な診断的産科処置は 52% 減少しました。著者らは、拡張NIPTアプローチがベルギーの出生前ケアにうまく導入されており、NIPTを提供する他の国々の枠組みとして機能する可能性があると結論付けている。
ゲノムワイドNIPSで検出された共通トリソミー以外の染色体異常の起源と臨床的関連性:TRIDENT研究の結果
オランダ
この出版物は、拡張 NIPT を使用して一般的な異数性以外の染色体異常を検出する潜在的な臨床的有用性を実証しました。この研究集団では、拡張 NIPT を受けた症例の 1.6% が、まれな常染色体トリソミーまたは大きな部分的欠失/重複である稀な染色体異常のスクリーニング陽性でした。これらの症例のうち、60% は、異常な胎児表現型や子宮内発育制限などの有害な妊娠転帰と関連していました。
ゲノムワイドな無細胞 DNA スクリーニング: コピー数変異に焦点を当てる
この出版物は、86,902 件の拡張 NIPT サンプルのコホートのうち、少なくとも 1 つの染色体部分欠失/重複のスクリーニング陽性となった 490 件 (0.56%) の症例に焦点を当てています。これらの症例の 50% で診断結果が得られ、PPV が 70% を超えることが判明しました。
拡大染色体疾患症候群に対する非侵襲的出生前スクリーニングの臨床的有用性
この出版物では、オールリスク妊娠集団における異数性およびゲノム全体の微小欠失/微小重複症候群の両方に対する拡張 NIPT の臨床成績を検証しました。単胎妊娠の患者94,085人のコホートが前向きに登録された。この拡張 NIPT では、サンプルの 1.2% で臨床的に重大な胎児染色体異常が検出され、スクリーニング陽性異常に対するそれぞれの PPV が計算されました。
よくある質問(FAQ)
A. 染色体の一部分が失われている状態を「部分欠失」、余分に増えている状態を「部分重複」といいます。その部位にある遺伝子の働きが変わることで、知的障害や発達の遅れ、身体的な特徴などがあらわれることがあります。失われる(増える)場所や大きさによって症状は異なります。
Q. NIPTで部分欠失・重複はどこまでわかりますか?
A. NIPTy Standardでは、常染色体44本のどの部位の欠失・重複も調べられます。東京衛生検査所(国内)の解析では、500万塩基(0.23%)以上の欠失・重複を報告対象としています。ただしNIPTは非確定的検査(スクリーニング)であり、確定診断には羊水検査が必要です。
Q. 検査で陽性となったら次に何をすればよいですか?
A. 陽性はそのまま診断ではありません。羊水検査で確定診断を行います。ヒロクリニックでは、他院での羊水検査にも使える費用サポート(プランに応じて最大10万〜30万円の補助)と、医師による遺伝カウンセリングをご用意しています。結果を一人で抱え込まず、まずはご相談ください。
Q. 部分欠失・重複はどのくらいの頻度で見つかりますか?
A. 検査集団によって幅がありますが、部分欠失・部分重複はおよそ0.1〜0.5%程度で報告されています。まれではあるものの、21・18・13トリソミー以外の染色体の変化も一定の割合で見つかります。あくまで可能性を調べる検査のため、詳しくは医師にご確認ください。
Q. どのプランを選べばよいですか?
A. 調べたい範囲やご希望に応じて、ベーシック・スタンダード・ワイド・プレミアムなどのプランがあります。ご自身に合うプランは、無料の「プラン診断」で数問に答えるだけで確認できます。迷ったときはLINEやお電話でもご相談いただけます。
Q. 結果はどのくらいで届きますか?費用は?
A. 東京衛生検査所(国内)で解析するため、最短2〜5日で結果をお届けします。費用は選ぶプランによって異なりますので、プラン診断またはお問い合わせでご確認ください。年齢制限や紹介状は不要で、心拍確認後の早い時期から受けられます。
日本皮膚科学会 皮膚科専門医/日本医師会 産業医/東京衛生検査所 指導監督医
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。
日本産科婦人科学会 産婦人科専門医/産婦人科(NIPT・出生前診断)
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。
日本産科婦人科学会 産婦人科専門医/産婦人科(NIPT・出生前診断)
この記事は、 ヒロクリニックNIPTの編集・監修体制 にもとづき、資格を持つ医師が内容を確認しています。


Copyright (c) NIPT Hiro Clinic All Rights Reserved.